Слова «раздача» морфологический и фонетический разбор
Объяснение правил деление (разбивки) слова «раздача» на слоги для переноса.
Онлайн словарь Soosle.ru поможет: фонетический и морфологический разобрать слово «раздача» по составу, правильно делить на слоги по провилам русского языка, выделить части слова, поставить ударение, укажет значение, синонимы, антонимы и сочетаемость к слову «раздача».
Содержимое:
- 1 Слоги в слове «раздача» деление на слоги
- 2 Как перенести слово «раздача»
- 3 Морфологический разбор слова «раздача»
- 4 Разбор слова «раздача» по составу
- 5 Синонимы слова «раздача»
- 6 Ударение в слове «раздача»
- 7 Фонетическая транскрипция слова «раздача»
- 8 Фонетический разбор слова «раздача» на буквы и звуки (Звуко-буквенный)
- 9 Предложения со словом «раздача»
- 10 Сочетаемость слова «раздача»
- 11 Значение слова «раздача»
- 12 Склонение слова «раздача» по подежам
- 13 Как правильно пишется слово «раздача»
- 14 Ассоциации к слову «раздача»
Слоги в слове «раздача» деление на слоги
Количество слогов: 3
По слогам: ра-зда-ча
По правилам школьной программы слово «раздача» можно поделить на слоги разными способами. Допускается вариативность, то есть все варианты правильные. Например, такой:
Допускается вариативность, то есть все варианты правильные. Например, такой:
раз-да-ча
По программе института слоги выделяются на основе восходящей звучности:
ра-зда-ча
Ниже перечислены виды слогов и объяснено деление с учётом программы института и школ с углублённым изучением русского языка.
з примыкает к этому слогу, а не к предыдущему, так как не является сонорной (непарной звонкой согласной)
Как перенести слово «раздача»
ра—здача
раз—дача
разда—ча
Морфологический разбор слова «раздача»
Часть речи:
Имя существительное
Грамматика:
часть речи: имя существительное;
одушевлённость: неодушевлённое;
род: женский;
число: единственное;
падеж: именительный;
отвечает на вопрос: (есть) Что?
Начальная форма:
раздача
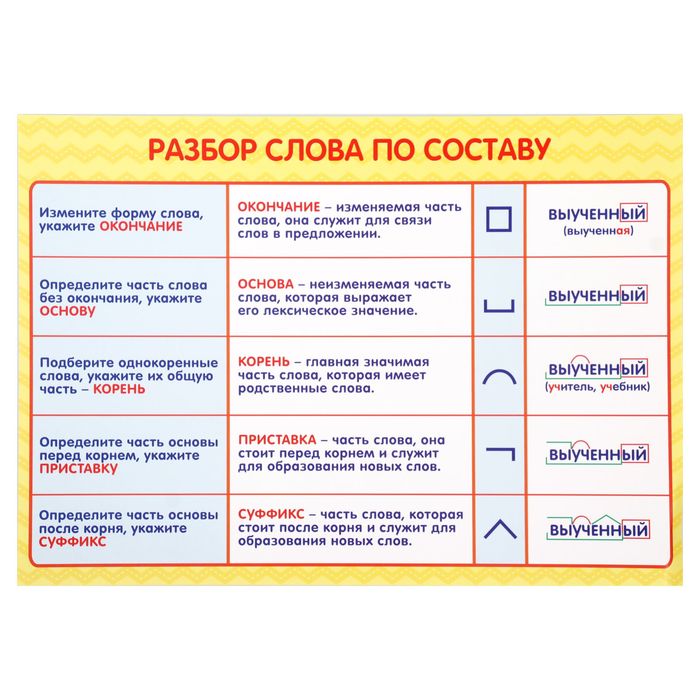
Разбор слова «раздача» по составу
| раз | приставка |
| да | корень |
| ч | суффикс |
| а | окончание |
раздача
Синонимы слова «раздача»
1. минираздача
минираздача
2. раздавание
3. кормораздача
4. сдача
Ударение в слове «раздача»
разда́ча — ударение падает на 2-й слог
Фонетическая транскрипция слова «раздача»
[разд`ач’а]
Фонетический разбор слова «раздача» на буквы и звуки (Звуко-буквенный)
| Буква | Звук | Характеристики звука | Цвет |
|---|---|---|---|
| р | [р] | согласный, звонкий непарный (сонорный), твёрдый | р |
| а | [а] | гласный, безударный | а |
| з | [з] | согласный, звонкий парный, твёрдый, шумный | з |
| д | [д] | согласный, звонкий парный, твёрдый, шумный | д |
| а | [`а] | гласный, ударный | а |
| ч | [ч’] | согласный, глухой непарный, мягкий, шипящий | ч |
| а | [а] | гласный, безударный | а |
Число букв и звуков:
На основе сделанного разбора делаем вывод, что в слове 7 букв и 7 звуков.
Буквы: 3 гласных буквы, 4 согласных букв.
Звуки: 3 гласных звука, 4 согласных звука.
Предложения со словом «раздача»
Никакие бесплатные раздачи хлеба из казны не помогали.
Источник: А. Р. Андреев, Как взять власть в России? Империя, ее народ и его охрана, 2011.
Это чтение завещания, а не раздача подарков.
Источник: А. Л. Сандер, Патримониум.
Плебс пользовался раздачами дарового хлеба.
Источник: И. В. Ткаченко, Всеобщая история в вопросах и ответах.
Сочетаемость слова «раздача»
1. денежные раздачи
2. в поместную раздачу
3. следующая раздача
4. раздача подарков
5. раздача пищи
6. раздача земель
7. к окну раздачи
8.
9. к стойке раздачи
10. попасть под раздачу
11. подойти к раздаче
12. начать раздачу
13. (полная таблица сочетаемости)
Значение слова «раздача»
РАЗДА́ЧА , -и, ж. Действие по знач. глаг. раздать1—раздавать1. Раздача денег. (Малый академический словарь, МАС)
Склонение слова «раздача» по подежам
| Падеж | Вопрос | Единственное числоЕд.ч. | Множественное числоМн.ч. |
|---|---|---|---|
| ИменительныйИм. | что? | раздача | раздачи |
| РодительныйРод. | чего? | раздачи | раздач |
| ДательныйДат. | чему? | раздаче | раздачам |
| ВинительныйВин. | что? | раздачу | раздачи |
| ТворительныйТв. | чем? | раздачей, раздачею | раздачами |
ПредложныйПред. | о чём? | раздаче | раздачах |
Как правильно пишется слово «раздача»
Орфография слова «раздача»Правильно слово пишется: разда́ча
Нумерация букв в слове
Номера букв в слове «раздача» в прямом и обратном порядке:
- 7
р
1 - 6
а
2 - 5
з
3 - 4
д
4 - 3
а
5 - 2
ч
6 - 1
а
7
Ассоциации к слову «раздача»
Милостыня
Автограф
Нуждающийся
Пряник
Земеля
Пайка
Бедняк
Листовка
Долгов
Пища
Жалованье
Туз
Повариха
Пособие
Подарок
Провиант
Презерватив
Приз
Казна
Продовольствие
Щедрость
Угощение
Хлеб
Суп
Пожертвование
Портик
Чина
Ссуда
Колода
Вотчина
Злоупотребление
Бачок
Празднество
Окошко
Чаевые
Награда
Зерно
Благотворительность
Покер
Брошюра
Коронация
Пир
Поощрение
Закупка
Распределение
Разбор
Землевладелец
Сувенир
Выдача
Сенат
Образцов
Паломничество
Корма
Порция
Помещик
Поверка
Столовая
Поднос
Жребий
Привилегия
Смотр
Стипендия
Загрузка
Котлета
Козырь
Комиссариат
Дворянство
Согражданин
Аристократия
Бесплатный
Слоновый
Хлебный
Щедрый
Благотворительный
Продуктовый
Денежный
Гуманитарный
Земельный
Продовольственный
Ежемесячный
Бездомный
Нищий
Съестной
Георгиевский
Праздничный
Пасхальный
Рекламный
Столовый
Раздавать
Заведовать
Раздать
Практиковаться
Организовывать
Сопровождаться
Производиться
Поспеть
Ведать
Поучаствовать
Приступить
Организовать
Значение слов в словарях
wordmap
Сложность и многогранность русского языка порой удивляют даже его носителей. Особенность заключается в отсутствии структурности. Ведь очень много вольностей допускается не только при построении предложений. Использование некоторых словоформ тоже имеет несколько вариаций.
Особенность заключается в отсутствии структурности. Ведь очень много вольностей допускается не только при построении предложений. Использование некоторых словоформ тоже имеет несколько вариаций.
Сложности и особенности работы со словом
В русском языке огромное количество допущений, которые нельзя встретить в других культурах. Ведь в речи часто используются не только литературные слова, которых свыше 150 тысяч. Но еще и диалектизмы. Так как в России много народов и культур, их более 250 тысяч. Неудивительно, что даже носителям языка иногда необходимо отыскать точные значения слов. Сделать это можно с помощью толковых словарей или специального сервиса WordMap.
Чем удобна такая площадка? Это понятный и простой словарь значений слов, использовать который предлагается в режиме онлайн. Сервис позволяет:
- узнать точное значение слова или идиомы;
- определить его корректное написание;
- понять, как правильно в нем ставить ударение.


Площадка предлагает ознакомиться с историей возникновения слова. Тут рассказывается, из какого языка или культуры оно пришло, когда и кем использовалось в речи.
Осуществляя поиск значения слов в словаре, важно понимать его суть. Ведь звуковая составляющая каждой лексической единицы в языке неразрывно связана с определенными предметами или явлениями. Вот почему при использовании сервиса не стоит ставить знак равенства между значением искомого слова и его понятием. Они связаны между собой, но не являются единым целым. К примеру, понятие слова «центр» можно определить как середину чего-либо. Однако конкретные значения могут указывать на внутреннюю часть комнаты, города, геометрической фигуры и т. д. Иногда речь идет о медицинской организации, математике или машиностроении. В многозначности и заключается сложность русского языка.
Поиск значений через WordMap
Для того, чтобы узнать, что значит слово, была проведена кропотливая работа. Ведь разные пособия и сборники могут давать разные значения одних и тех же лексических конструкций. Чтобы получить максимально полное представление о слове, стоит обратиться к сервису WordMap. В системе есть значения из наиболее популярных и авторитетных источников, включая словари:
Чтобы получить максимально полное представление о слове, стоит обратиться к сервису WordMap. В системе есть значения из наиболее популярных и авторитетных источников, включая словари:
- Ожегова;
- Даля;
- медицинского;
- городов;
- жаргонов;
- БСЭ и т. д.
Благодаря этому можно узнать не только все книжные, но и переносные значения лексической конструкции.
Только что искали:
хулы 3 секунды назад
дном 4 секунды назад
мазанкина 8 секунд назад
модаиипл 14 секунд назад
гром 15 секунд назад
графитируема 18 секунд назад
подчас 20 секунд назад
нигодява 21 секунда назад
скупать 23 секунды назад
ахом 24 секунды назад
миксер 28 секунд назад
асом 34 секунды назад
ларексв 43 секунды назад
аром 43 секунды назад
старая ушица 43 секунды назад
Ваша оценка
Закрыть
Спасибо за вашу оценку!
Закрыть
Последние игры в словабалдучепуху
| Имя | Слово | Угадано | Время | Откуда |
|---|---|---|---|---|
| Игрок 1 | сепараторщик | 0 слов | 18 минут назад | 178. 45.154.184 45.154.184 |
| Игрок 2 | ипнкрзда | 1 слово | 8 часов назад | 176.59.50.37 |
| Игрок 3 | апрапр | 2 слова | 13 часов назад | 51.15.48.52 |
| Игрок 4 | метасоматизм | 15 слов | 1 день назад | 176.59.124.156 |
| Игрок 5 | припоминание | 4 слова | 1 день назад | 95.71.47.218 |
| Игрок 6 | кот | 0 слов | 2 дня назад | 95.54.241.87 |
| Игрок 7 | город | 0 слов | 2 дня назад | 158.181.234.21 |
| Играть в Слова! | ||||
| Имя | Слово | Счет | Откуда | |
|---|---|---|---|---|
| Игрок 1 | шкала | 57:54 | 3 часа назад | 178. 205.243.188 205.243.188 |
| Игрок 2 | ловец | 56:52 | 3 часа назад | 178.205.243.188 |
| Игрок 3 | чабан | 60:53 | 3 часа назад | 178.205.243.188 |
| Игрок 4 | ворох | 59:59 | 3 часа назад | 178.205.243.188 |
| Игрок 5 | пашня | 43:47 | 4 часа назад | 178.205.243.188 |
| Игрок 6 | щиток | 54:55 | 4 часа назад | 176.59.122.48 |
| Игрок 7 | гайка | 56:62 | 4 часа назад | 176.59.122.48 |
| Играть в Балду! | ||||
| Имя | Игра | Вопросы | Откуда | |
|---|---|---|---|---|
| Вв | На одного | 20 вопросов | 14 часов назад | 178. 44.116.12 44.116.12 |
| Кетик | На одного | 10 вопросов | 1 день назад | 176.59.124.156 |
| Кот | На одного | 10 вопросов | 1 день назад | 176.59.124.156 |
| Lezina | На одного | 10 вопросов | 1 день назад | 185.61.7.232 |
| Соня | На одного | 10 вопросов | 1 день назад | 217.118.90.172 |
| Аниса | На одного | 10 вопросов | 2 дня назад | 5.128.126.42 |
| Соня | На одного | 10 вопросов | 2 дня назад | 5.128.126.42 |
| Играть в Чепуху! | ||||
морфология — Разбор по составу слова «задача»
Вопрос задан
Изменён 6 лет 9 месяцев назад
Просмотрен 11k раз
Каков морфологический состав слова «задача»? Учитывая его этимологию (задача <- задать <- дать), я пришёл к выводу, что тут приставка «за», корень «да», суффикс «ч» и окончание «а». Мне возразили так:
Мне возразили так:
Нельзя мотивировать слово «задача» от слова «дать». В слове «задача» диахронически произошло опрощение морфем, и морфемы, входящие в основу, по сути, слились в одну. Поэтому синхронически, с точки зрения состояния русского языка на данный момент, «задач» — это и есть один корень.
Верно ли это? Как узнать, слились ли морфемы в одну в других словах? Каковы признаки того, что слияние состоялось?
- морфология
3
В слове «задача» можно выделить мотивирующую основу ЗАДА, так как задать — задача. Но это вовсе не означает, что эта основа нечленимая, при разборе слова по составу она делится на приставку ЗА и корень ДА.
В лингвистике эта проблема называется ЧЛЕНИМОСТЬЮ основы слова, и различные школы имеют разный подход к ее решению. В общем случае можно выделить РАЗНЫЕ СТЕПЕНИ членимости. Максимальную степень членимости имеют слова с ДВУСТОРОННЕЙ СОПОСТАВИМОСТЬЮ (по корню и суффиксу): летчик — ЛЕТать и грузЧИК.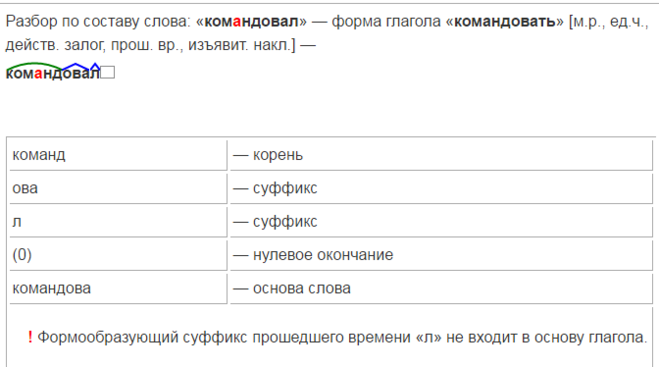 Сюда же относят слова со связанной основой: отпереть — запереть, включить — выключить. Меньшую степень членимости имеют основы с с ОДНОСТОРОННЕЙ СОПОСТАВИМОСТЬЮ (только по корню или только суффиксу): пасТУХ — летЧИК (по корню), бужеНИна — конИНа (по суффиксу) и т.д.
Сюда же относят слова со связанной основой: отпереть — запереть, включить — выключить. Меньшую степень членимости имеют основы с с ОДНОСТОРОННЕЙ СОПОСТАВИМОСТЬЮ (только по корню или только суффиксу): пасТУХ — летЧИК (по корню), бужеНИна — конИНа (по суффиксу) и т.д.
Таким образом, при разборе слова по составу мы не копируем словообразовательный разбор, а рассматриваем значения морфем, узнаваемые носителями на данном этапе развития языка. Слово «задача» имеет максимальную степень членимости: и по корню «задача — заДАть», и по суффиксу «задача — подаЧа, удаЧа. На современном этапе мы имеем приставку ЗА, корень ДА, суффикс Ч, окончание А, все они имеют узнаваемое значение. Суффикс Ч выделяется в существительных, обозначающих действие, названное по глаголу.
Материал дан по книге Николина Н.А. «современный русский язык. Морфемика»,2013, учебное пособие для филологических специальностей.
5
По разным словам распыляться не будем, слишком места мало в одном вопросе. Однокоренные к задаче слова: дача, отдача, удача, передача, сдача, выдача, подача, додача.
Однокоренные к задаче слова: дача, отдача, удача, передача, сдача, выдача, подача, додача.
Никто на сможет оспорить общий для всех слов корень ДА, ведь разные в них только приставки. Данный критерий достаточно нагляден.
Конкретно по задаче. Она задается для решения кому-то кем-то. Ученику, студенту, сотруднику, подчиненному и т.д. Кроме задачи, есть задание, в котором приставка и корень те же, а суффикс другой. То и другое слово произошли от глагола задать, который явно не имеет эксклюзивного морфологического состава. Приставка за- отлично выделяется в составе глаголов и существительных: задуть, забить, зажать, задумка, заплыв, забег, загиб, запил. Значение приставки: отдельное целенаправленное действие. Так что с морфологией все в порядке, она прозрачна и всем понятна. Корень ни в коем случае не слился с приставкой, это ошибочное, неправильное мнение.
В других словах может быть иная ситуация.
3
Да, верно: задач- вся основа — корень, потому что семантика слов дать, задать и задача разошлась.
Да/ть — вручить, предоставить, за/да/ть — поручить сделать, назначить, за/да/ни/е — то, что задали, поручили, а задач/а — то, что нужно исполнить, разрешить, появился новый оттенок трудности разрешения, связанный с мыслительной деятельностью. Дать задание и поставить задачу — смысл разный, хотя исторически от слова дать. (Ещё в 70-е годы у Тихонова было задача от задать), а вот в 80-е словари дают другой морфемный состав.
Признаки слияния — разные словообразовательные ряды: дать — дача (показаний) — выдача — данный — задание — задаток — задаточный — подача — податливый и т.д. Задача — задачка -сверхзадача — задачник.
Вопросы морфемного состава — трудная тема, в основном на чутье, но и ему доверять не стоит, только словарям, которые фиксируют изменения. Я 33 года проработала в школе, а как нужно выполнить для кого-то контрольную, обращаюсь к словарям. В основном на чутье, но частенько словарь всё же открываю: не изменилось ли что?
Да, верно: в словарях дается основа: задач. Но в то же время как в словаре Тихонова, так и других дается разбор по морфемам: за/да/ч/а. Не совсем понятно почему. Но я полностью согласен, что синхронически, с точки зрения русского языка, в этом слове выделяется только корень-основа.
Но в то же время как в словаре Тихонова, так и других дается разбор по морфемам: за/да/ч/а. Не совсем понятно почему. Но я полностью согласен, что синхронически, с точки зрения русского языка, в этом слове выделяется только корень-основа.
1
Вы извините, конечно, но все ваши рассуждения — философия чистой воды, равносильны гаданию на кофейной гуще. Можно сколько угодно долго размышлять и спорить, цвет лиловый или сиреневый, или как именно выглядели первые динозавры — ни на динозавров, ни на предмет это не подействует. Как верно заметил один из ораторов, правила придумали после того, как появились слова. Никого не хочу обидеть, но за уши можно притянуть что угодно. Соль не в этом. Я так понимаю, здесь собрались люди, в основном, с филологическим образованием, либо разбирающиеся. И если уж вы спорите, обращаетесь к справочникам, и справочники тоже спорят между собой, то почему детям в третьем классе дают такие задания и ставят потом 4 или 3 за неправильный морфемный разбор. Вопрос риторический. Непонятно, что система образования хочет этим добиться.
Вопрос риторический. Непонятно, что система образования хочет этим добиться.
Ваш ответ
Зарегистрируйтесь или войдите
Регистрация через Google
Регистрация через Facebook
Регистрация через почту
Отправить без регистрации
Почта
Необходима, но никому не показывается
Отправить без регистрации
Почта
Необходима, но никому не показывается
Нажимая на кнопку «Отправить ответ», вы соглашаетесь с нашими пользовательским соглашением, политикой конфиденциальности и политикой о куки
4 столпа визуализации данных: распределение, взаимосвязь, композиция, сравнение | by Mahbubul Alam
Фото Келли Тангай на Unsplash Много веков назад у ученых не было камер, чтобы делать снимки далеких галактик или крошечных бактерий под микроскопом. Рисунки были основным средством передачи наблюдений, идей и даже теорий. На самом деле способность рисовать абстрактные идеи и объекты была важным навыком для ученых (ознакомьтесь с коллекциями Уильяма Плейфэра 1700-х годов).
Рисунки были основным средством передачи наблюдений, идей и даже теорий. На самом деле способность рисовать абстрактные идеи и объекты была важным навыком для ученых (ознакомьтесь с коллекциями Уильяма Плейфэра 1700-х годов).
Это все еще так. Мы не можем фотографировать переменные распределения или их корреляцию. Вместо этого мы передаем их через рисунки и иллюстрации — то есть визуализацию данных — с помощью современных инструментов и технологий.
Набор данных содержит одну или несколько переменных, и мы можем визуализировать каждую из них и их взаимодействие с другими несколькими способами. Какую визуализацию выбрать, зависит от данных и типа информации, которую мы хотим передать. Однако, по сути, они бывают четырех разных видов:
- Распределение из единой переменной
- Отношения между двумя переменными
- Композиция из одной или множественной переменной
- Сравнение между разными категориями/индивидуумами
В этой статье, я будьте разбивая эти 4 краеугольных камня визуализации данных с помощью иллюстраций.
Важным понятием в статистике и науке о данных является распределение. Распределение обычно относится к вероятности возникновения результата. При распределении 100 бросков монеты сколько выпадет орел и сколько решка? Подобные частотные распределения представлены в виде гистограмм или кривых.
Ниже представлено распределение роста школьников в классе плавания. На оси X показаны разные категории роста, а на оси Y указано количество учащихся в каждой категории.
Частотное распределение роста учеников (рисунок: автор)Это частотное распределение. Но есть и другой тип распределения — более известный как дисперсия, — который показывает, как переменная рассеивается/распространяется по отношению к ее центральной тенденции.
Классическим представлением дисперсии является блочная диаграмма.
Разложение ящичковой диаграммы для отображения разброса значений переменной Приведенная выше ящичковая диаграмма представляет распределение количества авиапассажиров по субботам за несколько лет. Этот единственный график раскрывает так много информации — среднее/медианное количество пассажиров по субботам, минимумы и максимумы, выбросы и многое другое!
Этот единственный график раскрывает так много информации — среднее/медианное количество пассажиров по субботам, минимумы и максимумы, выбросы и многое другое!
В первые годы деревья становятся выше по мере взросления. Это связь между двумя переменными — ростом и возрастом.
рост = f(возраст)
В другом примере цена дома зависит от количества спальных мест, количества ванных комнат, местоположения, квадратных метров и т. д. Это отношение между одной зависимой и многими независимыми переменными.
цена = f(кровати, ванны, расположение, площадь)
Если рассматривать набор данных только как числа, то невозможно определить эти взаимосвязи. Но на самом деле можно, не вдаваясь в сложный статистический анализ, с помощью хорошей визуализации.
Соотношение возраста и роста класса учащихся Третьим краеугольным камнем визуализации данных является Сравнение. Этот тип визуального материала сравнивает несколько переменных в наборах данных или несколько категорий в рамках одной переменной.
Давайте посмотрим на следующие два изображения:
Изображения для сравненияНа изображении слева сравнивается переменная (зарплата) между двумя группами наблюдений (ученые и юристы) на гистограмме. Правая панель также представляет собой сравнительную диаграмму — в данном случае сравнение переменной (ВВП) между двумя группами (Великобритания и Канада), но во временном измерении.
Вы слышали о гистограммах с накоплением? Но я уверен, что вы знаете, что такое круговая диаграмма.
Целью этих диаграмм является отображение состава одной или нескольких переменных в абсолютных числах и в нормализованной форме (например, в процентах).
Диаграммы состава — это некоторые из методов визуализации старой школы, которые в настоящее время имеют ограниченные варианты использования (вам действительно нужна круговая диаграмма, чтобы показать состав желтых 10% и красных 15%?). Тем не менее, иногда они могут представить информацию визуально эстетично и привычно, в винтажном стиле.
Цель этой статьи состояла в том, чтобы обсудить четыре краеугольных камня визуализации данных: распределение, взаимосвязь, сравнение и композиция. Прежде чем изучать инструменты и методы визуализации, важно понять, какова цель визуализации и какую информацию вы хотите передать. В будущих статьях я буду писать о конкретных инструментах на языках программирования Python и R, включая matplotlib 9.0068 , сибон и ggplot2 . Следите за обновлениями!
[Примечание: все рисунки, кроме первого, нарисованы автором]
полевой справочник для композиционного анализа данных any-omics | GigaScience
Abstract
Background
Секвенирование нового поколения (NGS) позволило определить последовательность и относительное содержание всех нуклеотидов в биологическом образце или образце из окружающей среды. Краеугольным камнем NGS является количественная оценка присутствия РНК или ДНК в виде подсчетов. Однако эти подсчеты не являются подсчетами сами по себе: их величина определяется произвольно глубиной секвенирования, а не входным материалом. Следовательно, подсчеты должны пройти нормализацию перед использованием. Обычные методы нормализации требуют ряда допущений: они предполагают, что большинство признаков неизменны и что все исследуемые среды имеют одинаковую пропускную способность для синтеза нуклеотидов. Эти предположения часто не поддаются проверке и могут оказаться неверными при сравнении разнородных выборок.
Краеугольным камнем NGS является количественная оценка присутствия РНК или ДНК в виде подсчетов. Однако эти подсчеты не являются подсчетами сами по себе: их величина определяется произвольно глубиной секвенирования, а не входным материалом. Следовательно, подсчеты должны пройти нормализацию перед использованием. Обычные методы нормализации требуют ряда допущений: они предполагают, что большинство признаков неизменны и что все исследуемые среды имеют одинаковую пропускную способность для синтеза нуклеотидов. Эти предположения часто не поддаются проверке и могут оказаться неверными при сравнении разнородных выборок.
Результаты
Методы, разработанные в области композиционного анализа данных, предлагают общее решение, которое не требует предположений и справедливо для всех данных. Здесь мы обобщаем существующую литературу, чтобы предоставить краткое руководство о том, как применять анализ композиционных данных к данным подсчета NGS.
Выводы
Подчеркивая ограничения общего размера библиотеки, эффективного размера библиотеки и нормализации всплесков, мы предлагаем преобразование логарифмического отношения в качестве общего решения для ответа на вопрос: «Относительно некоторой важной активности клетки, что меняется?»
Введение
Появление секвенирования нового поколения (NGS) позволило ученым беспрецедентно исследовать биологические системы. За постоянно уменьшающуюся сумму денег можно определить последовательность и относительное содержание всех нуклеотидных фрагментов в образце [1]. NGS работает путем секвенирования популяции фрагментов ДНК, включая изоляты РНК с обратной транскрипцией. Помимо общего использования для обнаружения вариантов и сборки генома, NGS используется для количественной оценки относительного содержания (i) видов РНК в тканях (секвенирование РНК [RNA-Seq]) [1], (ii) разнообразия организмов из окружающей среды ( метагеномика) [2], (iii) виды РНК из окружающей среды (мета-транскриптомика) [3] и (iv) области генома, на которые нацелен белок (секвенирование иммунопреципитации хроматина) [4], среди прочего. В последнее время усовершенствования в протоколах секвенирования позволили проводить эти измерения на уровне одной клетки, при этом РНК-Seq одной клетки является наиболее зрелой технологией. В большинстве приложений используется аналогичная процедура, при которой ДНК или РНК выделяются из образцов, при необходимости фильтруются по размеру или другим свойствам [5], преобразуются в библиотеку комплементарных ДНК (кДНК) фрагментов нуклеотидов, секвенируются на секвенаторе, а затем сопоставляются с эталоном.
За постоянно уменьшающуюся сумму денег можно определить последовательность и относительное содержание всех нуклеотидных фрагментов в образце [1]. NGS работает путем секвенирования популяции фрагментов ДНК, включая изоляты РНК с обратной транскрипцией. Помимо общего использования для обнаружения вариантов и сборки генома, NGS используется для количественной оценки относительного содержания (i) видов РНК в тканях (секвенирование РНК [RNA-Seq]) [1], (ii) разнообразия организмов из окружающей среды ( метагеномика) [2], (iii) виды РНК из окружающей среды (мета-транскриптомика) [3] и (iv) области генома, на которые нацелен белок (секвенирование иммунопреципитации хроматина) [4], среди прочего. В последнее время усовершенствования в протоколах секвенирования позволили проводить эти измерения на уровне одной клетки, при этом РНК-Seq одной клетки является наиболее зрелой технологией. В большинстве приложений используется аналогичная процедура, при которой ДНК или РНК выделяются из образцов, при необходимости фильтруются по размеру или другим свойствам [5], преобразуются в библиотеку комплементарных ДНК (кДНК) фрагментов нуклеотидов, секвенируются на секвенаторе, а затем сопоставляются с эталоном. для количественной оценки относительного изобилия. Поскольку все данные получены из одного и того же анализа, можно было бы ожидать, что они будут подвергаться одинаковому анализу. Однако это неверно: скорее, методы, адаптированные для одного типа данных, не распространяются на другой (например, методы RNA-Seq имеют завышенные показатели ложных открытий [FDR] при применении к метагеномным данным [6,7]).
для количественной оценки относительного изобилия. Поскольку все данные получены из одного и того же анализа, можно было бы ожидать, что они будут подвергаться одинаковому анализу. Однако это неверно: скорее, методы, адаптированные для одного типа данных, не распространяются на другой (например, методы RNA-Seq имеют завышенные показатели ложных открытий [FDR] при применении к метагеномным данным [6,7]).
Fernandes et al. утверждал, что анализ всех данных NGS может быть концептуально унифицирован путем признания композиционного характера этих данных [8]. Под «композиционным» мы подразумеваем, что распространенность любого 1-нуклеотидного фрагмента можно интерпретировать только относительно другого. Это свойство возникает из самого секвенсора; секвенсор по своей конструкции может секвенировать только фиксированное количество нуклеотидных фрагментов. Следовательно, конечное количество секвенированных фрагментов ограничено произвольным пределом, так что удвоение входного материала не удваивает общее количество отсчетов. Это ограничение также означает, что увеличение присутствия любого 1-нуклеотидного фрагмента обязательно снижает наблюдаемое количество всех других транскриптов [9].] и применяется как к массовым данным, так и к данным секвенирования отдельных клеток. Это особенно проблематично при сравнении клеток, которые производят больше общей РНК, чем их компаратор (например, клетки с высоким уровнем c-Myc, которые активируют 90% всех транскриптов без соразмерного подавления [10]). Однако, даже если бы секвенатор мог непосредственно секвенировать каждую молекулу РНК внутри клетки, сами клетки являются составными из-за ограничений по объему и энергии, которые ограничивают синтез РНК, о чем свидетельствует наблюдение, что более мелкие клетки одного типа содержат пропорционально меньше общего количества мессенджеров. РНК (мРНК) [11].
Это ограничение также означает, что увеличение присутствия любого 1-нуклеотидного фрагмента обязательно снижает наблюдаемое количество всех других транскриптов [9].] и применяется как к массовым данным, так и к данным секвенирования отдельных клеток. Это особенно проблематично при сравнении клеток, которые производят больше общей РНК, чем их компаратор (например, клетки с высоким уровнем c-Myc, которые активируют 90% всех транскриптов без соразмерного подавления [10]). Однако, даже если бы секвенатор мог непосредственно секвенировать каждую молекулу РНК внутри клетки, сами клетки являются составными из-за ограничений по объему и энергии, которые ограничивают синтез РНК, о чем свидетельствует наблюдение, что более мелкие клетки одного типа содержат пропорционально меньше общего количества мессенджеров. РНК (мРНК) [11].
Композиционные данные несут только относительную информацию. Следовательно, они существуют в симплексном пространстве с размерностью на 1 меньше, чем компоненты.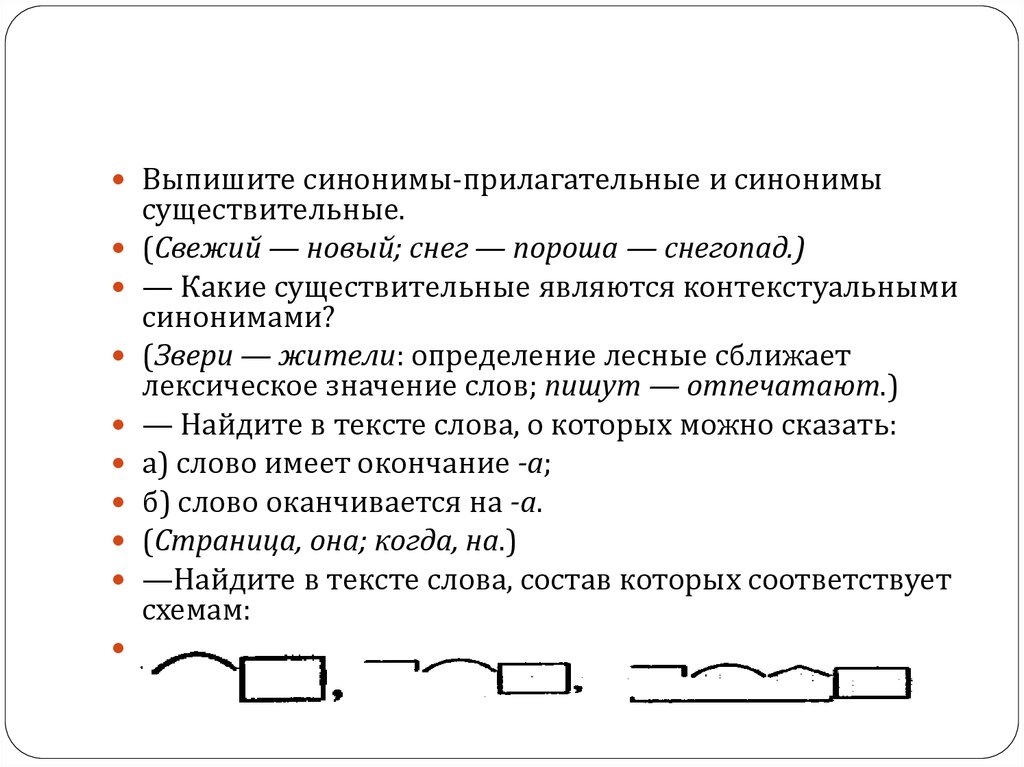 Анализ относительных данных, как если бы они были абсолютными, может привести к ошибочным результатам для нескольких распространенных методов [12–14] (также продемонстрировано в дополнительном анализе S1). Во-первых, статистические модели, которые предполагают независимость между признаками, ошибочны из-за взаимной зависимости между компонентами [15]. Во-вторых, расстояния между образцами вводят в заблуждение и хаотично чувствительны к произвольному включению или исключению компонентов [16]. В-третьих, компоненты могут оказаться окончательно коррелированными, даже если они статистически независимы [17]. По этим причинам данные о составе создают особые проблемы для анализа дифференциальной экспрессии, кластеризации и корреляционного анализа, обычно применяемого к данным NGS, а также к другим данным, которые измеряют относительное содержание малых молекул (например, данные спектрометрических пиков [18]). Для композиционных данных NGS каждый образец называется «композицией», а каждый вид нуклеотидов называется «компонентом» [13,14].
Анализ относительных данных, как если бы они были абсолютными, может привести к ошибочным результатам для нескольких распространенных методов [12–14] (также продемонстрировано в дополнительном анализе S1). Во-первых, статистические модели, которые предполагают независимость между признаками, ошибочны из-за взаимной зависимости между компонентами [15]. Во-вторых, расстояния между образцами вводят в заблуждение и хаотично чувствительны к произвольному включению или исключению компонентов [16]. В-третьих, компоненты могут оказаться окончательно коррелированными, даже если они статистически независимы [17]. По этим причинам данные о составе создают особые проблемы для анализа дифференциальной экспрессии, кластеризации и корреляционного анализа, обычно применяемого к данным NGS, а также к другим данным, которые измеряют относительное содержание малых молекул (например, данные спектрометрических пиков [18]). Для композиционных данных NGS каждый образец называется «композицией», а каждый вид нуклеотидов называется «компонентом» [13,14].
Существует 3 основных подхода к анализу композиционных данных. Во-первых, «зависимый от нормализации» подход стремится нормализовать данные, чтобы восстановить абсолютное содержание. Однако нормализация зависит от предположений, которые могут быть неверны вне строго контролируемых экспериментов. Например, популярные методы нормализации RNA-Seq предполагают, что большинство транскрипты имеют одинаковое абсолютное содержание в образцах [19,20], предположение, которое не выполняется для вышеупомянутых клеток с высоким содержанием c-Myc [10]. Во-вторых, «зависимый от трансформации» подход преобразует данные в отношении эталона сделать статистические выводы относительно выбранного эталона [12]. В-третьих, «независимый от преобразований» подход выполняет расчеты непосредственно по компонентам [21] или отношениям компонентов [22]9.0003
Последние 2 подхода представляют собой композиционный анализ данных (CoDA). В отличие от методов, основанных на нормализации, методы CoDA обобщают все данные, относительные или абсолютные. В этой статье мы описываем унифицированный конвейер для анализа данных подсчета NGS, все части которого полностью способны моделировать неопределенность подсчетов с низкой численностью. Во-первых, мы показываем, как существующие программные инструменты CoDA можно использовать для получения композиционно обоснованных и биологически значимых выводов. Во-вторых, мы показываем, как эти методы могут соответствовать сложному дизайну исследования, облегчать анализ горизонтально интегрированных мультиомных данных и приспосабливаться к приложениям машинного обучения. В-третьих, мы показываем, как композиционность может систематически искажать результаты, если ее игнорировать. Наконец, мы заканчиваем обсуждением ключевых проблем, связанных с нормализацией всплесков, и показываем, как структура CoDA применяется конкретно к данным секвенирования одной клетки.
В этой статье мы описываем унифицированный конвейер для анализа данных подсчета NGS, все части которого полностью способны моделировать неопределенность подсчетов с низкой численностью. Во-первых, мы показываем, как существующие программные инструменты CoDA можно использовать для получения композиционно обоснованных и биологически значимых выводов. Во-вторых, мы показываем, как эти методы могут соответствовать сложному дизайну исследования, облегчать анализ горизонтально интегрированных мультиомных данных и приспосабливаться к приложениям машинного обучения. В-третьих, мы показываем, как композиционность может систематически искажать результаты, если ее игнорировать. Наконец, мы заканчиваем обсуждением ключевых проблем, связанных с нормализацией всплесков, и показываем, как структура CoDA применяется конкретно к данным секвенирования одной клетки.
Методы
Обзор пайплайна
Наш пайплайн использует бесплатные программные инструменты для языка программирования R. Он начинается с ненормализованной «матрицы подсчета», созданной в результате выравнивания и картирования чтения библиотеки последовательностей. Детали, касающиеся контроля качества, сборки, выравнивания и картирования чтения, выходят за рамки этой статьи и широко освещались в других местах (например, [23,24]). Эта матрица подсчета записывает, сколько раз каждый признак (например, расшифровка или операционная таксономическая единица [OTU]) появляется в каждом образце. Большинство программ возвращает измерения в виде целых чисел, хотя некоторые используют непрерывные значения (например, квазисчета лосося [25]) или другую пропорциональную единицу (например, количество транскриптов на миллион [TPM] [26]). Для многих методов CoDA единицы не имеют значения. Однако небольшие подсчеты несут больше неопределенности, чем большие подсчеты, и наш конвейер может моделировать это напрямую. Поэтому мы рекомендуем использовать нескорректированные «необработанные подсчеты». TPM также можно использовать с методами CoDA, но это может привести к смещению при моделировании малых количеств, если размер библиотеки сильно различается между образцами.
Он начинается с ненормализованной «матрицы подсчета», созданной в результате выравнивания и картирования чтения библиотеки последовательностей. Детали, касающиеся контроля качества, сборки, выравнивания и картирования чтения, выходят за рамки этой статьи и широко освещались в других местах (например, [23,24]). Эта матрица подсчета записывает, сколько раз каждый признак (например, расшифровка или операционная таксономическая единица [OTU]) появляется в каждом образце. Большинство программ возвращает измерения в виде целых чисел, хотя некоторые используют непрерывные значения (например, квазисчета лосося [25]) или другую пропорциональную единицу (например, количество транскриптов на миллион [TPM] [26]). Для многих методов CoDA единицы не имеют значения. Однако небольшие подсчеты несут больше неопределенности, чем большие подсчеты, и наш конвейер может моделировать это напрямую. Поэтому мы рекомендуем использовать нескорректированные «необработанные подсчеты». TPM также можно использовать с методами CoDA, но это может привести к смещению при моделировании малых количеств, если размер библиотеки сильно различается между образцами. В противном случае данные не должны подвергаться дальнейшей нормализации или стандартизации и никогда не должны содержать отрицательных значений. На рис. 1 представлена схема нашего унифицированного конвейера NGS.
В противном случае данные не должны подвергаться дальнейшей нормализации или стандартизации и никогда не должны содержать отрицательных значений. На рис. 1 представлена схема нашего унифицированного конвейера NGS.
Рисунок 1:
Открыть в новой вкладкеСкачать слайд
На этом рисунке показано, как наш унифицированный конвейер NGS может вписаться в более крупный рабочий процесс. Цветные прямоугольники обозначают процедуры, применимые к любому относительному набору данных. Оранжевым цветом обозначены необязательные действия по удалению и изменению нуля, представленные в разделе «Обработка нуля». Зеленым цветом мы описываем методы, зависящие от преобразования логарифмического отношения, представленные в разделе «Анализ, зависящий от преобразования». Это включает дифференциальный анализ изобилия отдельных признаков и анализ пропорциональности пар признаков. Желтым цветом мы описываем независимые от трансформации методы, представленные в разделе «Анализ, независимый от трансформации». Сюда входит анализ различий в средних логарифмических отношениях пар признаков. Серым цветом мы описываем другие важные шаги, уникальные для изучаемого типа данных, но не рассматриваемые здесь. w.r.t.: в отношении.
Сюда входит анализ различий в средних логарифмических отношениях пар признаков. Серым цветом мы описываем другие важные шаги, уникальные для изучаемого типа данных, но не рассматриваемые здесь. w.r.t.: в отношении.
Сбор данных
Чтобы продемонстрировать полезность нашего проекта, мы используем общедоступные данные о динамике РНК и белка, экспрессируемых дендритными клетками мыши после воздействия липополисахарида (ЛПС), мощного иммуногенного стимулятора. Данные RNA-Seq и масс-спектрометрии (МС) были получены уже предварительно обработанными для измерения относительной распространенности 3147 генов в единицах, эквивалентных TPM [27]. Данные RNA-Seq и MS содержали 28 перекрывающихся образцов, охватывающих 2 состояния с 7 временными точками и 2 повторениями для каждого.
# Чтение данных RNA-Seq
rnaseq <- read.csv("rnaseq-x.csv", row.names=1)
rnaseq.annot <- read.csv("rnaseq-y.csv ", row.names=1)
# Чтение данных Mass Spec HL
masshl <- read. csv("masshl-x.csv", row.names=1)
csv("masshl-x.csv", row.names=1)
masshl.annot <- read.csv("masshl-y.csv", row.names=1)
# Мы создадим подмножество Mass Spec, чтобы включить временные точки
# с соответствующим измерением RNA-Seq
# (используется в «Вертикальной интеграции данных»)
в РНК и МС <- masshl.annot$Time
mashl <- masshl[ в РНК и МС]
masshl.annot <- masshl.annot[в РНК и МС,]
8 New
Представляя этот рабочий процесс, мы проводим новый анализ данных Йовановича и др. [27], чтобы узнать, как содержание транскриптов мРНК и содержание белка изменяются в ответ на стимуляцию ЛПС.Это включает анализ относительного дифференциального содержания, анализ координации ген-ген и анализ дифференциальной координации ген-ген. Кроме того, мы объединяем 2 типа данных с дифференциальным анализом пропорциональности, чтобы оценить, как стехиометрия мРНК отличается от стехиометрии белка в ответ на обработку ЛПС. В отличие от исходного анализа, представленного Йованович и др. [27], мы не используем нормализацию числа транскриптов на миллион (TPM). Скорее, мы утверждаем, что TPM переделывают уже составной набор данных как еще один композиционный набор данных. et (только с другим знаменателем). В дополнительном анализе S1 мы показываем, как TPM вносят систематические ошибки. Это связано с тем, что, когда ссылка не выбрана явно, произвольная ссылка все еще неявно присутствует. Мы также включили приложение (Дополнительный анализ S2), в котором оценивается, как несколько процедур обработки нуля влияют на анализ пропорциональности и дифференциальной пропорциональности.
[27], мы не используем нормализацию числа транскриптов на миллион (TPM). Скорее, мы утверждаем, что TPM переделывают уже составной набор данных как еще один композиционный набор данных. et (только с другим знаменателем). В дополнительном анализе S1 мы показываем, как TPM вносят систематические ошибки. Это связано с тем, что, когда ссылка не выбрана явно, произвольная ссылка все еще неявно присутствует. Мы также включили приложение (Дополнительный анализ S2), в котором оценивается, как несколько процедур обработки нуля влияют на анализ пропорциональности и дифференциальной пропорциональности.
Добавление программного обеспечения
Этот рабочий процесс в основном использует 3 пакета программного обеспечения с открытым исходным кодом, все из которых доступны для языка программирования R. К ним относятся zCompositions [28], ALDEx2 [8,29] и propr [30, 31]. Читатель может загрузить эти программные пакеты с сайтов Bioconductor и CRAN.
install.packages("zCompositions")
install.packages("propr")
install.packages("BiocManager")
# Читать ‘::’ как ‘функция установки’ из
BiocManager package"
BiocManager::install("ALDEx2")
библиотека (zCompositions)
библиотека (ALDEx2)
библиотека (propr)
При подготовке этого рабочего процесса мы внесли несколько вкладов в композиционные данные Вселенная программного обеспечения для анализа. Во-первых, мы представляем новую функцию propr::aldex2propr, которая интегрирует пакеты ALDEx2 и propr, вычисляя средний коэффициент пропорциональности по экземплярам Монте-Карло, сгенерированным ALDEx2. Во-вторых, мы представляем новую функцию propr::updateCutoffs, которая переставляет FDR через различные пороговые значения коэффициента пропорциональности. В-третьих, мы представляем функцию propr::propd, которая реализует метод дифференциальной пропорциональности, описанный Erb et al. [31], включая реализацию процедуры обработки нуля на основе преобразования Бокса-Кокса. Эти новые дополнения делают возможным полный рабочий процесс композиционного анализа данных.
Эти новые дополнения делают возможным полный рабочий процесс композиционного анализа данных.
Контрольная проверка
Хотя можно разработать «нормализующую» ссылку, используя набор допущений, мы предпочитаем альтернативную структуру, которая не требует какой-либо нормализации. Мы используем эту структуру, потому что она обеспечивает более общее решение для анализа данных - omics. Таким образом, предлагаемый нами рабочий процесс можно использовать для анализа объемной РНК-Seq, одноклеточной РНК-Seq, метагеномики, метаболомики, липидомики и других данных.
Хотя представленные здесь программные инструменты не нормализуют данные, их можно сравнить с обычными методами, приняв допущение, что явная ссылка выполняет своего рода «логарифмическую нормализацию отношения». В этих условиях ALDEx2 может с высокой точностью идентифицировать дифференциальную численность в данных RNA-Seq [8,32] и контролировать частоту ложноположительных результатов в очень разреженных данных метагеномного подсчета 16S [6]. Между тем, было показано, что анализ пропорциональности превосходит все 15 конкурирующих мер ассоциации в задачах кластеризации отдельных ячеек и сетевых выводов по 213 наборам данных [33]. Хотя дифференциальный анализ пропорциональности еще не был протестирован, он формально связан с дисперсионным анализом, основным тестом в большинстве биологических исследований. В качестве статистического теста на значимость он действителен везде, где действителен дисперсионный анализ. Мы также включили приложение (Дополнительный анализ S2), в котором оценивается, как несколько процедур обработки нуля влияют на анализ пропорциональности и дифференциальной пропорциональности.
Между тем, было показано, что анализ пропорциональности превосходит все 15 конкурирующих мер ассоциации в задачах кластеризации отдельных ячеек и сетевых выводов по 213 наборам данных [33]. Хотя дифференциальный анализ пропорциональности еще не был протестирован, он формально связан с дисперсионным анализом, основным тестом в большинстве биологических исследований. В качестве статистического теста на значимость он действителен везде, где действителен дисперсионный анализ. Мы также включили приложение (Дополнительный анализ S2), в котором оценивается, как несколько процедур обработки нуля влияют на анализ пропорциональности и дифференциальной пропорциональности.
Обработка нулей
Общие стратегии обработки нулей
Методы CoDA зависят от логарифмов, которые не вычисляются для нулей. Следовательно, мы должны обращаться к нулям до или во время конвейера. Прежде чем обращаться с нулями, аналитик должен сначала рассмотреть природу нулей. Существует 3 типа нулей: (i) «округление», также называемое «выборкой», когда признак существует в образце ниже предела обнаружения; (ii) «подсчет», когда признак существует в выборке, но подсчет не является достаточно исчерпывающим, чтобы увидеть его хотя бы один раз; и (iii) «существенный», когда признак вообще не существует в выборке [34]. Подход к обработке нулей зависит от природы нулей [34]. Для данных NGS нуклеотидный фрагмент либо секвенирован, либо нет, и не будет содержать округляющих нулей. Поскольку не существует общей методологии работы с существенными нулями в строгой структуре CoDA [34], мы предполагаем, что любой признак, присутствующий в ≥1 выборке, может появиться в другой выборке, если секвенировать с бесконечной глубиной, и, таким образом, рассматривать все нули NGS как «счетчик». нули». Другие также предположили, что существенные нули данных подсчета NGS в достаточной степени моделируются как нули выборки [35].
Прежде чем обращаться с нулями, аналитик должен сначала рассмотреть природу нулей. Существует 3 типа нулей: (i) «округление», также называемое «выборкой», когда признак существует в образце ниже предела обнаружения; (ii) «подсчет», когда признак существует в выборке, но подсчет не является достаточно исчерпывающим, чтобы увидеть его хотя бы один раз; и (iii) «существенный», когда признак вообще не существует в выборке [34]. Подход к обработке нулей зависит от природы нулей [34]. Для данных NGS нуклеотидный фрагмент либо секвенирован, либо нет, и не будет содержать округляющих нулей. Поскольку не существует общей методологии работы с существенными нулями в строгой структуре CoDA [34], мы предполагаем, что любой признак, присутствующий в ≥1 выборке, может появиться в другой выборке, если секвенировать с бесконечной глубиной, и, таким образом, рассматривать все нули NGS как «счетчик». нули». Другие также предположили, что существенные нули данных подсчета NGS в достаточной степени моделируются как нули выборки [35].
Существует 2 основных подхода к работе с нулями. При «удалении признаков» компоненты с нулями исключаются, что дает подкомпозицию, которую можно анализировать любым методом CoDA. Удаление признака обычно уместно, когда признак содержит много нулей и всегда может быть оправдан для существенных нулей. В «модификации функции» нули заменяются ненулевым значением, с изменением или без изменения ненулевых значений. Аналитики могут выбрать одну или обе процедуры обработки нулей, но всегда должны демонстрировать, что удаление или модификация функций, загруженных нулями, не меняет общей интерпретации результатов.
Модификация функции с помощью zCompositions
Для «нулей счета» Martin-Fernández et al. рекомендуют заменять нули байесовско-мультипликативной стратегией замены, сохраняющей соотношения между ненулевыми компонентами [34], реализованной в пакете zCompositions в виде функции cmultRepl [28]. В качестве альтернативы можно использовать мультипликативную стратегию простой замены, при которой нули заменяются фиксированным значением <1 композиционно устойчивым образом. Здесь мы используем zCompositions для замены нулей.
Здесь мы используем zCompositions для замены нулей.
#Стандартные функции ожидают строк в качестве образцов
#, поэтому мы будем транспонировать матрицу
RNASEQ <-T (RNASEQ)
MASSHL <-T (MASSHL)
#Теперь можем заменить нуа
# опция "p-counts" имеет функцию return
# псевдосчетчики вместо пропорций
Можно интерпретировать эту «повышенную регуляцию» как означающую, что ген увеличивает свою экспрессию в ответ на стимуляцию LPS больше, чем ядерный фактор κB (NFκB). Все P -значения соответствуют ожиданиям скорректированных по Бенджамини-Хохбергу P -значений, рассчитанных на основе t -теста Уэлча по 128 смоделированным экземплярам данных. Выбирая ссылку, относящуюся к изучаемой биологической системе, мы можем получить значимую информацию из данных без какой-либо необходимости нормализации. В таблице 1 межгрупповые различия — это различия между двумя состояниями (определены для каждого случая Дирихле), внутригрупповые различия — это максимальное различие между экземплярами Дирихле (определено для каждого состояния), а величины эффекта — это отношение между -групповые различия до максимума внутригрупповых различий (определены для каждого экземпляра Дирихле). Столбцы «Величина эффекта», «Разница (между)» и «Разница (внутри)» сообщают о медианной величине эффекта, медианной разнице между группами и медианной разнице внутри группы соответственно.
Столбцы «Величина эффекта», «Разница (между)» и «Разница (внутри)» сообщают о медианной величине эффекта, медианной разнице между группами и медианной разнице внутри группы соответственно.
masshl.no0 <- cmultRepl(masshl, output = "p-counts'')
Многие инструменты композиционного программного обеспечения имеют собственные встроенные процедуры обработки нуля. Хотя zCompositions не обязательно лучше этих встроенных процедур, мы понимаем, что прямое удаление нулей имеет практическое преимущество: используя zCompositions в сочетании с логарифмическим преобразованием, аналитики могут сразу же применять большинство обычных анализов к своим композиционным данным. Поскольку zCompositions позволяет читателям использовать методы помимо представленных здесь, мы решили включить его в качестве первой части нашего полевого руководства. Тем не менее, мы рекомендуем читателям ознакомиться с дополнительным анализом S2, в котором оценивается, как несколько процедур обработки нуля влияют на анализ пропорциональности и дифференциальной пропорциональности.
Анализ, зависящий от преобразования
Преобразование логарифмического отношения
Все компоненты в композиции являются взаимозависимыми свойствами, которые нельзя понять по отдельности. Поэтому любой анализ отдельных компонентов делается относительно эталона. Эта ссылка превращает каждую выборку в неограниченное пространство, где можно использовать любой статистический метод. Преобразование центрированного логарифмического отношения (clr) использует среднее геометрическое вектора выборки в качестве эталона [36]. Аддитивное преобразование логарифмического отношения (alr) использует один компонент в качестве эталона [36]. Другие преобразования используют специализированные ссылки, основанные на среднем геометрическом подмножества компонентов (все вместе называемые мультиаддитивными преобразованиями логарифмического отношения [malr] [32]). Одним из преобразований malr является преобразование межквартильного логарифмического отношения (iqlr), в котором используются компоненты в межквартильном диапазоне дисперсии [37]. Другое, надежное центрированное преобразование логарифмического отношения (rclr), использует только ненулевые компоненты [38].
Другое, надежное центрированное преобразование логарифмического отношения (rclr), использует только ненулевые компоненты [38].
Важно отметить, что преобразования не являются нормализацией: в то время как нормализации утверждают, что преобразуют данные в абсолютном выражении, преобразования этого не делают. Результаты анализа, основанного на трансформации, должны интерпретироваться по отношению к выбранному эталону. Из них наиболее распространено преобразование clr:
$$\begin{eqnarray}\textrm{clr}(\mathbf {x}_j) = \left[\ln \frac{x_{1,j}}{g (\mathbf {x}_j)},...,\ln \frac{x_{D,j}}{g(\mathbf {x}_j)}\right],\end{eqnarray}$$
(1)
, где |$\mathbf{x}_j$| — это j -я выборка, а |$g(\mathbf {x}_j)$| является его средним геометрическим. Другие преобразования заменяют |$g(\mathbf {x}_j)$| с другой ссылкой.
Преобразование изометрического логарифмического отношения (ILR) использует ортонормированный базис в качестве эталона [39] и предпочтительнее, когда требуется невырожденная ковариационная матрица [21]. Когда основой является ветвь дендрограммы, ilr предлагает интуитивно понятный способ сопоставления одного набора компонентов с другим набором компонентов. Эти контрасты, называемые балансами, использовались для анализа метагеномных данных на основе эволюционных деревьев [40, 41], но их можно было бы применять к любым данным, если бы было доступно дерево с аналогичным значением.
Когда основой является ветвь дендрограммы, ilr предлагает интуитивно понятный способ сопоставления одного набора компонентов с другим набором компонентов. Эти контрасты, называемые балансами, использовались для анализа метагеномных данных на основе эволюционных деревьев [40, 41], но их можно было бы применять к любым данным, если бы было доступно дерево с аналогичным значением.
Каждое преобразование подразумевает свою собственную ссылку(и). В большинстве практических случаев выбор преобразования будет зависеть от предпочтительной интерпретации. Анализ данных clr покажет, как гены (или OTU) ведут себя по отношению к среднему показателю для выборки. Анализ данных alr и malr покажет, как гены (или OTU) ведут себя по отношению к 1 или более явно выбранным внутренним ссылкам. Анализ данных iqlr покажет, как гены (или OTU) ведут себя по отношению к межквартильному («надежному») среднему значению для каждой выборки. В композиционной структуре ни одна из них не является нормализацией: каждая новая переменная представляет собой логарифмическое отношение исходной переменной, деленное на эталон, и поэтому должна интерпретироваться как своего рода логарифмическая разница внутри выборки. Хотя разница между преобразованием и нормализацией может показаться тонкой, она может оказать глубокое влияние на выводы, сделанные в результате анализа. Хотя соблазн будет существовать, никогда нельзя путать преобразованные данные с абсолютным изобилием.
Хотя разница между преобразованием и нормализацией может показаться тонкой, она может оказать глубокое влияние на выводы, сделанные в результате анализа. Хотя соблазн будет существовать, никогда нельзя путать преобразованные данные с абсолютным изобилием.
Дифференциальный анализ численности с помощью ALDEx2
Дифференциальный анализ численности (DA) призван определить, какие признаки различаются по численности между экспериментальными группами. Пакет ALDEx2 тестирует DA в композиционных данных, выполняя одномерный статистический анализ данных, преобразованных в логарифмическом отношении [8,29]. Он делает это с помощью уровня сложности, который контролирует технические вариации, находя ожидание B смоделированных экземпляров данных, каждый из которых выбран из распределения Дирихле. Эта процедура неявно моделирует неопределенность низких значений, а также обрабатывает нули.
Важно отметить, что ALDEx2 идентифицирует DA относительно выбранной ссылки. По умолчанию эта ссылка представляет собой среднее геометрическое композиции. Возможно, если не вероятно, что средние центры не являются идеальными точками отсчета; если это так, то различия в преобразованных количествах не будут отражать различия в абсолютных количествах. С другой стороны, если можно предположить, что выбранный эталон действительно имеет фиксированную абсолютную численность во всех образцах, то преобразование логарифмического отношения может быть оценено как «нормализация логарифмического отношения» [14]. В этих условиях ALDEx2 может с высокой точностью идентифицировать DA в данных RNA-Seq [8,32] и контролировать частоту ложноположительных результатов в очень разреженных данных метагеномного подсчета 16S [6]. Однако интерпретация «нормализации логарифмического отношения» подразумевает аналогичное предположение, подразумеваемое другими инструментами DA: большинство видов транскриптов остаются неизменными [42]. В качестве альтернативы можно выбрать произвольный эталон, основанный на биологической гипотезе, для определения «относительной DA», даже если эталон не имеет фиксированного содержания в образцах.
По умолчанию эта ссылка представляет собой среднее геометрическое композиции. Возможно, если не вероятно, что средние центры не являются идеальными точками отсчета; если это так, то различия в преобразованных количествах не будут отражать различия в абсолютных количествах. С другой стороны, если можно предположить, что выбранный эталон действительно имеет фиксированную абсолютную численность во всех образцах, то преобразование логарифмического отношения может быть оценено как «нормализация логарифмического отношения» [14]. В этих условиях ALDEx2 может с высокой точностью идентифицировать DA в данных RNA-Seq [8,32] и контролировать частоту ложноположительных результатов в очень разреженных данных метагеномного подсчета 16S [6]. Однако интерпретация «нормализации логарифмического отношения» подразумевает аналогичное предположение, подразумеваемое другими инструментами DA: большинство видов транскриптов остаются неизменными [42]. В качестве альтернативы можно выбрать произвольный эталон, основанный на биологической гипотезе, для определения «относительной DA», даже если эталон не имеет фиксированного содержания в образцах. На рис. 2 показано, как выбранная ссылка меняет интерпретацию DA.
На рис. 2 показано, как выбранная ссылка меняет интерпретацию DA.
Рисунок 2:
Открыть в новой вкладкеСкачать слайд
На этом рисунке показано, как интерпретация дифференциальной численности зависит от выбранного эталона. На левом поле мы показываем логарифмическое содержание 3 генов (RPL19, FSCN1 и IL1B) для обработанных ЛПС клеток (оранжевый) и контроля (синий). Для композиционных данных эти изобилия не имеют смысла сами по себе, потому что ограниченная сумма накладывает «предвзятость закрытия». На верхнем поле мы показываем логарифмическое количество двух ссылок: среднее геометрическое выборок (а-ля clr) и основанную на гипотезе ссылку NFκB (а-ля alr). В середине мы показываем изобилие логарифмического отношения функции левого поля, деленного на ссылку верхнего поля (эквивалент левого поля минус верхнее поле в логарифмическом пространстве). РПЛ19один кажется более обильным в контроле, но фактически имеет эквивалентную экспрессию по сравнению со средним геометрическим; однако он имеет значительно более высокую экспрессию в контроле по сравнению с NFκB. С другой стороны, один FSCN1, по-видимому, более экспрессируется в клетках, обработанных LPS, что остается верным при сравнении со средним геометрическим; однако он имеет эквивалентную экспрессию относительно NFκB (интерпретируется как экспрессия NFκB и FSCN1, изменяющаяся сходным образом в ответ на стимуляцию LPS). Сам по себе IL1B экспрессируется более высоко в обработанных LPS клетках, что остается верным при сравнении со средним геометрическим и с NFκB (интерпретируется как экспрессия IL1B, которая становится даже выше, чем экспрессия NFκB в ответ на стимуляцию LPS). Выбор эталона делает нормализацию ненужной, но требует изменения интерпретации.
С другой стороны, один FSCN1, по-видимому, более экспрессируется в клетках, обработанных LPS, что остается верным при сравнении со средним геометрическим; однако он имеет эквивалентную экспрессию относительно NFκB (интерпретируется как экспрессия NFκB и FSCN1, изменяющаяся сходным образом в ответ на стимуляцию LPS). Сам по себе IL1B экспрессируется более высоко в обработанных LPS клетках, что остается верным при сравнении со средним геометрическим и с NFκB (интерпретируется как экспрессия IL1B, которая становится даже выше, чем экспрессия NFκB в ответ на стимуляцию LPS). Выбор эталона делает нормализацию ненужной, но требует изменения интерпретации.
Для запуска ALDEx2 пользователь должен предоставить данные счетчика с целочисленными значениями, вектор групповых меток и ссылку. Ссылка может быть «все» (для clr), «iqlr» (для iqlr) или одна или несколько указанных пользователем функций (для alr или malr). Здесь мы используем среднее геометрическое 2 субъединиц NFκB в качестве эталона, основанного на гипотезе, выбранном потому, что LPS активирует NFκB для контроля транскрипции других иммунных генов [43].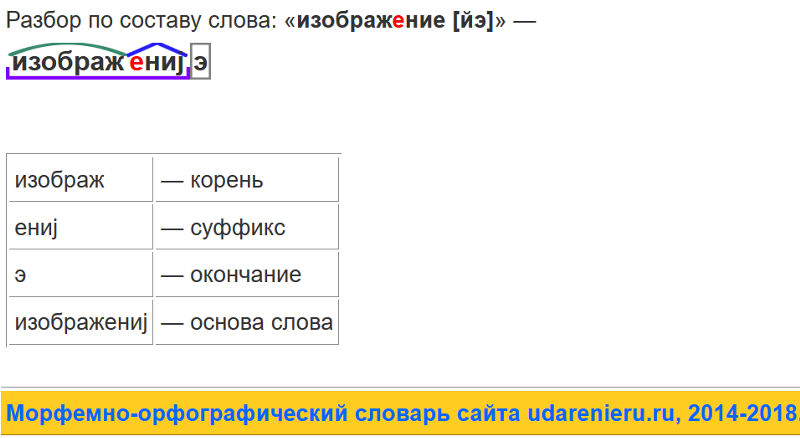 С этой ссылкой повышающая регуляция означает, что экспрессия гена увеличивается по сравнению с экспрессией NFκB, что обеспечивает четкую биологическую интерпретацию. В таблице 1 перечислены 47 генов с повышенной активностью по сравнению с NFκB.
С этой ссылкой повышающая регуляция означает, что экспрессия гена увеличивается по сравнению с экспрессией NFκB, что обеспечивает четкую биологическую интерпретацию. В таблице 1 перечислены 47 генов с повышенной активностью по сравнению с NFκB.
Таблица 1.
47 генов, выбранных как значительно активизированные с помощью ALDEx2 при использовании субъединиц NFκB в качестве эталона
| Ген . | Размер эффекта . | Разница (между) . | Разница (внутри) . | Ожидаемый Бенджамини-Хохберг P -значение . | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| II1b | 4,7372 | 3.9576 | 0.6912 | 0.0000 | ||||||
| Irg1 | 4.3462 | 3.8904 | 0.7888 | 0.0000 | ||||||
| Il1a | 3.5950 | 3.8242 | 0. 9037 9037 | 0.0000 | ||||||
| Cd40 | 2,2887 | 5,3325 | 2,0422 | 0,0000 | ||||||
| IFIH2 | 2,2056 | 2,8529 | 999988 2,20562,8529 | 99999988888892,8529 | 999999988888889999992,8529 | 9999888889,2056.0289 | 0.0000 | |||
| Isg15 | 1.9678 | 4.4490 | 1.8330 | 0.0000 | ||||||
| Oasl1 | 1.9304 | 5.6562 | 2.1200 | 0.0000 | ||||||
| Ifit1 | 1.8317 | 5.6101 | 2,0773 | 0,0000 | ||||||
| Ptgs2 | 1,6923 | 4,0869 | 2,0606 | 0,000 | 9999992,0606.0271 | Gbp5;Gbp1 | 1.6523 | 2.4494 | 1.2349 | 0. 0000 0000 |
| Rsad2 | 1.4933 | 6.2747 | 2.4692 | 0.0001 | ||||||
| Marcksl1 | 1.4886 | 1.0748 | 0.5740 | 0.0001 | ||||||
| BC006779 | 1.4686 | 2.2184 | 1.2465 | 0.0001 | ||||||
| Mndal | 1.4163 | 2.1047 | 1.5182 | 0.0000 | ||||||
| Parp14 | 1.3139 | 1.7655 | 0.9357 | 0.0002 | ||||||
| Ifi205 | 1.2916 | 5.3159 | 3.4587 | 0.0026 | ||||||
| Slc7a2 | 1,2883 | 1,3797 | 0,9920 | 0,0002 | ||||||
| Ifit2 | 2.6744 | 0.0002 | ||||||||
| Clic4 | 1.2037 | 0.8486 | 0.5765 | 0.0003 | ||||||
| Sp140 | 1. 1612 1612 | 1.0030 | 0.7385 | 0.0005 | ||||||
| Cmpk2 | 1.1149 | 5,7323 | 2,1088 | 0,0003 | ||||||
| STAT5A | 1,0806 | 0,8666 | 06 0,646119689 | 0,8666 | 806111111689 | 0,8666 | 8806.1689 | 0,8666 | .0289 | 0.0017 |
| Ifi47 | 1.0443 | 2.0495 | 1.5704 | 0.0030 | ||||||
| Pyhin1 | 1.0152 | 1.9150 | 1.4752 | 0.0024 | ||||||
| Ifit3 | 0.9978 | 4.7313 | 3.2116 | 0.0012 | ||||||
| Ccl5 | 0.9962 | 2.0765 | 1.6671 | 0.0015 | ||||||
| Acsl1 | 0.9937 | 1.0837 | 1.0073 | 0.0009 | ||||||
| Il1rn | 0.9811 | 0. 6795 6795 | 0.6366 | 0.0017 | ||||||
| Irgm1 | 0.9755 | 1.7076 | 1.0634 | 0.0094 | ||||||
| IIGP;Iigp1 | 0,9588 | 3,5610 | 3,1760 | 0,0023 | ;02890.9541 | 1.2867 | 1.0478 | 0.0041 | ||
| Daxx | 0.9118 | 1.1938 | 0.9013 | 0.0119 | ||||||
| Flnb | 0.8639 | 1.6654 | 1.8185 | 0.0122 | ||||||
| CD274 | 0,8299 | 0,6050 | 0,6354 | 0,0051 | ||||||
| TREX1 | 0,81111119999999988888888888888888888888888888888,0 | 8888888888888888888 гг.0289 | 0.6350 | 0.0090 | ||||||
| Car13 | 0.7586 | 1.1455 | 1.2839 | 0.0140 | ||||||
| Xaf1 | 0. 7550 7550 | 1.5118 | 1.4338 | 0.0214 | ||||||
| Gbp3 | 0.7478 | 1.5118 | 1.4837 | 0.0128 | ||||||
| Ehd1 | 0.7460 | 0.3648 | 0.4812 | 0.0078 | ||||||
| Gm4902 | 0.7413 | 1.9614 | 1.7899 | 0.0151 | ||||||
| Rasa4 | 0.7254 | 0.8805 | 0.9109 | 0.0478 | ||||||
| Oas3 | 0.7089 | 1.5673 | 1.7756 | 0,0213 | ||||||
| SERPINB2 | 0,7048 | 1,7770 | 2,1734 | 0,0272 | ; 0,0272 | |||||
| 88;0289 | 0.6947 | 1.4875 | 1.6956 | 0.0425 | ||||||
| Gbp2 | 0.6597 | 1.5376 | 1.7339 | 0.0212 | ||||||
| Saa3 | 0. 6291 6291 | 1.0259 | 1.5384 | 0.0187 | ||||||
| SBDS | 0,5522 | 0,3107 | 0,5363 | 0,0443 |
 3325
3325  2465
2465 0,
0288 0.0002
 8666
8666 0288 1.0152
 013
013  7089
7089 One can интерпретируйте эту «повышенную регуляцию» как означающую, что ген увеличивает свою экспрессию в ответ на стимуляцию LPS больше, чем NFκB. Все P -значения соответствуют ожиданиям скорректированного Бенджамини-Хохберга P — значения, рассчитанные на основе теста Уэлча t для 128 смоделированных экземпляров данных. Выбирая ссылку, относящуюся к изучаемой биологической системе, мы можем получить значимую информацию из данных без какой-либо необходимости нормализации. В этой таблице межгрупповые различия — это различия между двумя состояниями (определены для каждого случая Дирихле), внутригрупповые различия — это максимальное различие между экземплярами Дирихле (определено для каждого условия), а величины эффекта — это отношение между -групповые различия до максимума внутригрупповых различий (определены для каждого экземпляра Дирихле). Столбцы «Величина эффекта», «Разница (между)» и «Разница (внутри)» сообщают о медианной величине эффекта, медианной разнице между группами и медианной разнице внутри группы соответственно.
Выбирая ссылку, относящуюся к изучаемой биологической системе, мы можем получить значимую информацию из данных без какой-либо необходимости нормализации. В этой таблице межгрупповые различия — это различия между двумя состояниями (определены для каждого случая Дирихле), внутригрупповые различия — это максимальное различие между экземплярами Дирихле (определено для каждого условия), а величины эффекта — это отношение между -групповые различия до максимума внутригрупповых различий (определены для каждого экземпляра Дирихле). Столбцы «Величина эффекта», «Разница (между)» и «Разница (внутри)» сообщают о медианной величине эффекта, медианной разнице между группами и медианной разнице внутри группы соответственно.
Открыть в новой вкладке
Таблица 1.
47 генов, выбранных как значительно активизированные ALDEx2 при использовании субъединиц NFκB в качестве эталона
| Ген . | Размер эффекта
. | Разница (между) . | Разница (внутри) . | Ожидаемый Бенджамини-Хохберг P -значение . | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Il1b | 4.7372 | 3.9576 | 0.6912 | 0.0000 | |||||||
| Irg1 | 4.3462 | 3.8904 | 0.7888 | 0.0000 | |||||||
| Il1a | 3.5950 | 3.8242 | 0.9037 | 0.0000 | |||||||
| Cd40 | 2.2887 | 5.3325 | 2.0422 | 0.0000 | |||||||
| Ifih2 | 2.2056 | 2.8529 | 1.1157 | 0.0000 | |||||||
| Isg15 | 1.9678 | 4.4490 | 1.8330 | 0.0000 | |||||||
| Oasl1 | 1.9304 | 5.6562 | 2.1200 | 0.0000 | |||||||
| Ifit1 | 1,8317 | 5,6101 | 2,0773 | 0,0000 | |||||||
| Ptgs223 | 4. 0869 0869 | 2.0606 | 0.0002 | ||||||||
| Gbp5;Gbp1 | 1.6523 | 2.4494 | 1.2349 | 0.0000 | |||||||
| Rsad2 | 1.4933 | 6.2747 | 2.4692 | 0.0001 | |||||||
| MARCKSL1 | 1,4886 | 1,0748 | 0,5740 | 0,0001 | |||||||
| BC006779 | 1,4686 | BC006779 | 8686888888888888888888888889 гг.0289 | 1.2465 | 0.0001 | ||||||
| Mndal | 1.4163 | 2.1047 | 1.5182 | 0.0000 | |||||||
| Parp14 | 1.3139 | 1.7655 | 0.9357 | 0.0002 | |||||||
| Ifi205 | 1.2916 | 5,3159 | 3,4587 | 0,0026 | |||||||
| Slc7a2 | 1,2883 | 1,3797 9 | 9 9,0288 | 0.0002 | |||||||
| Ifit2 | 1.2292 | 5. 4975 4975 | 2.6744 | 0.0002 | |||||||
| Clic4 | 1.2037 | 0.8486 | 0.5765 | 0.0003 | |||||||
| Sp140 | 1.1612 | 1.0030 | 0.7385 | 0.0005 | |||||||
| Cmpk2 | 1.1149 | 5.7323 | 2.1088 | 0.0003 | |||||||
| Stat5a | 1.0806 | 0.8666 | 0.6461 | 0.0017 | |||||||
| Ifi47 | 1.0443 | 2.0495 | 1.5704 | 0.0030 | |||||||
| Pyhin1 | 1.0152 | 1.9150 | 1.4752 | 0.0024 | |||||||
| Ifit3 | 0,9978 | 4,7313 | 3,2116 | 0,0012 | |||||||
| Ccl89 962 | 2.0765 | 1.6671 | 0.0015 | ||||||||
| Acsl1 | 0.9937 | 1.0837 | 1.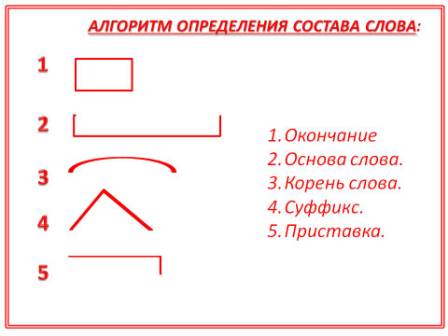 0073 0073 | 0.0009 | |||||||
| Il1rn | 0.9811 | 0.6795 | 0.6366 | 0.0017 | |||||||
| Irgm1 | 0,9755 | 1,7076 | 1,0634 | 0,0094 | |||||||
| IIGP;Iigp1 | 3 0, 289 | 3.1760 | 0.0023 | ||||||||
| Rnf213;AK217856 | 0.9541 | 1.2867 | 1.0478 | 0.0041 | |||||||
| Daxx | 0.9118 | 1.1938 | 0.9013 | 0.0119 | |||||||
| Flnb | 0.8639 | 1.6654 | 1.8185 | 0.0122 | |||||||
| Cd274 | 0.8299 | 0.6050 | 0.6354 | 0.0051 | |||||||
| Trex1 | 0.8171 | 0.5647 | 0.6350 | 0.0090 | |||||||
| Car13 | 0.7586 | 1.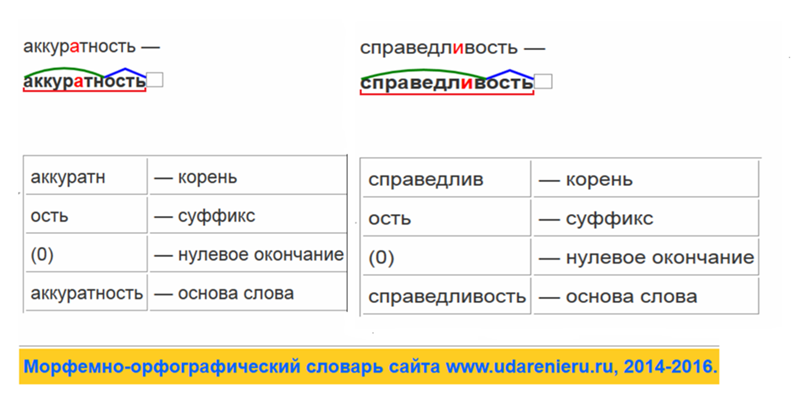 1455 1455 | 1.2839 | 0.0140 | |||||||
| Xaf1 | 0.7550 | 1.5118 | 1,4338 | 0,0214 | |||||||
| GBP3 | 0,7478 | 1,5118 | 1,4837 | 0,0128 | 1,4837 | 0,0128 | Ehd1 | 0.7460 | 0.3648 | 0.4812 | 0.0078 |
| Gm4902 | 0.7413 | 1.9614 | 1.7899 | 0.0151 | |||||||
| Rasa4 | 0.7254 | 0.8805 | 0.9109 | 0.0478 | |||||||
| Oas3 | 0,7089 | 1,5673 | 1,7756 | 0,0213 | |||||||
| 1.7770 | 2. 1734 1734 | 0.0272 | |||||||||
| Dhx58;D11lgp2 | 0.6947 | 1.4875 | 1.6956 | 0.0425 | |||||||
| Gbp2 | 0.6597 | 1.5376 | 1.7339 | 0.0212 | |||||||
| Saa3 | 0.6291 | 1.0259 | 1.5384 | 0.0187 | |||||||
| Sbds | 0.5522 | 0.3107 | 0,5363 | 0,0443 |
| Джин . | Размер эффекта . | Разница (между) . | Разница (внутри) . | Ожидаемый Бенджамини-Хохберг P -значение . | |||||
|---|---|---|---|---|---|---|---|---|---|
| Il1b | 4,7372 | 3,9576 | 0,6912 | 0,0000 | |||||
| Irg1 | 4.3462 | 3.8904 | 0.7888 | 0.0000 | |||||
| Il1a | 3. 5950 5950 | 3.8242 | 0.9037 | 0.0000 | |||||
| Cd40 | 2.2887 | 5.3325 | 2.0422 | 0,0000 | |||||
| Ifih2 | 2,2056 | 2,8529 | 1,1157 | 0,0000 9 12 | 02891.9678 | 4.4490 | 1.8330 | 0.0000 | |
| Oasl1 | 1.9304 | 5.6562 | 2.1200 | 0.0000 | |||||
| Ifit1 | 1.8317 | 5.6101 | 2.0773 | 0.0000 | |||||
| Ptgs2 | 1,6923 | 4,0869 | 2,0606 | 0,0002 | |||||
| Gbp5; Gbp1 | 9 9,02889 | 2.4494 | 1.2349 | 0.0000 | |||||
| Rsad2 | 1.4933 | 6.2747 | 2.4692 | 0.0001 | |||||
| Marcksl1 | 1.4886 | 1.0748 | 0.5740 | 0. 0001 0001 | |||||
| BC006779 | 1.4686 | 2,2184 | 1,2465 | 0,0001 | |||||
| Мндал | 1,4163 | 2,10487 2,104870288 1.51820.0000 | |||||||
| Parp14 | 1.3139 | 1.7655 | 0.9357 | 0.0002 | |||||
| Ifi205 | 1.2916 | 5.3159 | 3.4587 | 0.0026 | |||||
| Slc7a2 | 1.2883 | 1.3797 | 0,9920 | 0,0002 | |||||
| IFIT2 | 1,2292 | 5,4975 | 2,6744 | 9880282,6744 | 988.10282,6744 | 988.9752,6744 | 988.9752,6744 | 99752,6744 | 9975.0289 |
| Clic4 | 1.2037 | 0.8486 | 0.5765 | 0.0003 | |||||
| Sp140 | 1.1612 | 1.0030 | 0. 7385 7385 | 0.0005 | |||||
| Cmpk2 | 1.1149 | 5.7323 | 2.1088 | 0,0003 | |||||
| Stat5a | 1,0806 | 0,8666 | 0,6461 | 0,0017 4028 | 9017 402802891.0443 | 2.0495 | 1.5704 | 0.0030 | |
| Pyhin1 | 1.0152 | 1.9150 | 1.4752 | 0.0024 | |||||
| Ifit3 | 0.9978 | 4.7313 | 3.2116 | 0.0012 | |||||
| Ccl5 | 0.9962 | 2.0765 | 1.6671 | 0.0015 | |||||
| Acsl1 | 0.9937 | 1.0837 | 1.0073 | 0.0009 | |||||
| Il1rn | 0.9811 | 0.6795 | 0.6366 | 0.0017 | |||||
| Irgm1 | 0.9755 | 1.7076 | 1.0634 | 0. 0094 0094 | |||||
| IIGP;Iigp1 | 0.9588 | 3.5610 | 3.1760 | 0.0023 | |||||
| Rnf213;AK217856 | 0.9541 | 1.2867 | 1.0478 | 0.0041 | |||||
| Daxx | 0.9118 | 1.1938 | 0.9013 | 0.0119 | |||||
| Flnb | 0.8639 | 1.6654 | 1.8185 | 0.0122 | |||||
| Cd274 | 0.8299 | 0.6050 | 0.6354 | 0.0051 | |||||
| Trex1 | 0.8171 | 0.5647 | 0.6350 | 0.0090 | |||||
| Car13 | 0.7586 | 1.1455 | 1.2839 | 0.0140 | |||||
| Xaf1 | 0.7550 | 1.5118 | 1.4338 | 0.0214 | |||||
| Gbp3 | 0.7478 | 1.5118 | 1.4837 | 0. 0128 0128 | |||||
| Ehd1 | 0.7460 | 0.3648 | 0.4812 | 0.0078 | |||||
| Gm4902 | 0.7413 | 1.9614 | 1.7899 | 0.0151 | |||||
| Rasa4 | 0.7254 | 0.8805 | 0.9109 | 0.0478 | |||||
| Oas3 | 0.7089 | 1.5673 | 1.7756 | 0.0213 | |||||
| Serpinb2 | 0,7048 | 1,7770 | 2,1734 | 0,0272 | |||||
| 0289 | 1.6956 | 0.0425 | |||||||
| Gbp2 | 0.6597 | 1.5376 | 1.7339 | 0.0212 | |||||
| Saa3 | 0.6291 | 1.0259 | 1.5384 | 0.0187 | |||||
| Sbds | 0.5522 | 0,3107 | 0,5363 | 0,0443 |
Можно интерпретировать эту «активацию» как означающую, что ген увеличивает свою экспрессию в ответ на стимуляцию ЛПС больше, чем NFκB. Все P -значения соответствуют ожиданиям скорректированных по Бенджамини-Хохбергу P -значений, рассчитанных на основе t -теста Уэлча по 128 смоделированным экземплярам данных. Выбирая ссылку, относящуюся к изучаемой биологической системе, мы можем получить значимую информацию из данных без какой-либо необходимости нормализации. В этой таблице межгрупповые различия — это различия между двумя состояниями (определены для каждого случая Дирихле), внутригрупповые различия — это максимальное различие между экземплярами Дирихле (определено для каждого условия), а величины эффекта — это отношение между -групповые различия до максимума внутригрупповых различий (определены для каждого экземпляра Дирихле). Столбцы «Величина эффекта», «Разница (между)» и «Разница (внутри)» сообщают о медианной величине эффекта, медианной разнице между группами и медианной разнице внутри группы соответственно.
Все P -значения соответствуют ожиданиям скорректированных по Бенджамини-Хохбергу P -значений, рассчитанных на основе t -теста Уэлча по 128 смоделированным экземплярам данных. Выбирая ссылку, относящуюся к изучаемой биологической системе, мы можем получить значимую информацию из данных без какой-либо необходимости нормализации. В этой таблице межгрупповые различия — это различия между двумя состояниями (определены для каждого случая Дирихле), внутригрупповые различия — это максимальное различие между экземплярами Дирихле (определено для каждого условия), а величины эффекта — это отношение между -групповые различия до максимума внутригрупповых различий (определены для каждого экземпляра Дирихле). Столбцы «Величина эффекта», «Разница (между)» и «Разница (внутри)» сообщают о медианной величине эффекта, медианной разнице между группами и медианной разнице внутри группы соответственно.
Открыть в новой вкладке
#Давайте использовать подразделения NFKB в качестве ссылки ALR
Ref <-grep («NFKB», Colnames (RNASEQ))
#ALDEX2 ожидает:
#
# 'условия': результат эксперимента
# 'деном': эталон преобразования логарифмического отношения , "ЛПС"))
tt <- aldex(reads = t(ceiling(rnaseq)),
условия = условия,
деном = ref)
# ALDEx2 outputs #2Hawedata. frame:
frame:
. значение p с поправкой на FDR
# 'эффект': размер эффекта
# Ниже мы получаем названия генов
# с относительно большим количеством
# в группе LPS 90tt005-ttb.b000h
9000h [tt$we.eBH < .05,]up <- rownames(tt.bh05[tt.bh05$effect > 0,])
Анализ пропорциональности с помощью propr
Анализ пропорциональности предназначен для выявления координации признаков в композиционных данных [44,45] без предположения о разреженности в сети ассоциаций [46,47]. Пакет propr проверяет наличие согласованности признаков во всех образцах, независимо от метки группы, вычисляя 1 из 3 показателей пропорциональности. Было показано, что два из них превосходят все 15 конкурирующих мер ассоциации в задачах кластеризации отдельных ячеек и сетевых выводов по 213 наборам данных [33]. Мера по умолчанию, ρ p напоминает корреляцию в том смысле, что она находится в диапазоне [−1, 1]. Как и DA, анализ пропорциональности требует ссылки.
Как и DA, анализ пропорциональности требует ссылки.
# proprexpects:
# 'counts': матрица данных со строками в качестве выборок
# 'metric': метрика пропорциональности для расчета )
pr <- propr(counts = rnaseq.no0,
metric = "rho",
ivar = "clr")
Пакет propr предлагает 2 альтернативы нулевой обработке. Функция propr::aldex2propr вычисляет ожидаемую пропорциональность на основе смоделированных экземпляров, сгенерированных ALDEx2, что опять-таки устраняет неопределенность низких значений [48]. Альфа-аргумент будет использовать процедуру обработки нулей, основанную на преобразовании Бокса-Кокса, прагматическом подходе, который допускает существенные нули, но не подпадает под строгие рамки CoDA [49]. Преобразование Бокса-Кокса с α = 0,5, по-видимому, хорошо работает при моделировании (см. Дополнительный анализ S2). Для пропорциональности параметр 9 не рассчитываем0041 P -значения. Вместо этого мы переставляем FDR для данного порога. Исходя из этого, мы выбираем отсечку ρ p > 0,45, чтобы контролировать FDR ниже 5%. Виньетка пакета описывает несколько встроенных инструментов для визуализации пропорциональности. На рис. 3 показаны выходные данные функции getNetwork.
Исходя из этого, мы выбираем отсечку ρ p > 0,45, чтобы контролировать FDR ниже 5%. Виньетка пакета описывает несколько встроенных инструментов для визуализации пропорциональности. На рис. 3 показаны выходные данные функции getNetwork.
Рисунок 3:
Открыть в новой вкладкеСкачать слайд
Сеть, границы которой указывают на высокий уровень координации между экспрессией генов по отношению к среднему геометрическому для каждого образца. Цвет узла указывает на дифференциальную экспрессию относительно NFκB. Связи между красными узлами указывают на гены, экспрессия которых скоординировано увеличивается больше, чем NFκB. Связи между белыми узлами указывают на гены, экспрессия которых увеличивается в той же степени, что и NFκB, скоординированным образом. Связи между синими узлами указывают на гены, экспрессия которых либо (а) активирует меньше, чем NFκB, (б) не изменяется абсолютно, или (в) подавляет экспрессию, все скоординировано. Высокий уровень связности между всеми узлами предполагает сильную скоординированную реакцию на LPS. Как и коррелированные пары, пропорциональные пары могут иметь любой наклон в нелогарифмическом пространстве. Обратите внимание, что эта сеть показывает только хорошо скоординированные события (где ρ р > 0,9).
Как и коррелированные пары, пропорциональные пары могут иметь любой наклон в нелогарифмическом пространстве. Обратите внимание, что эта сеть показывает только хорошо скоординированные события (где ρ р > 0,9).
# Мы можем выбрать хорошую отсечку для 'rho'
# путем перестановки FDR в различных отсечках
# Ниже мы используем [0, .05, ..., .95, 1]
pr <- updateCutoffs(pr, cutoff = seq(0, 1, .05))
pr@fdr
# Давайте визуализируем, используя строгое отсечение
getNetwork(pr, cutoff = 0,90, 0.9, get col1 = 03 Results pr, cutoff = 0,9)
Пропорциональность зависит от преобразования логарифмического отношения и должна интерпретироваться по отношению к выбранному эталону. Хотя пропорциональность кажется более устойчивой к ложным ассоциациям, чем корреляция [30, 44], ошибочное предположение о том, что эталон имеет фиксированное абсолютное содержание во всех образцах, может привести к неверным выводам [45]. Мы интерпретируем пропорциональность на основе clr как обозначение координации, которая следует общей тенденции данных. Другими словами, эти пропорциональные гены движутся вместе как индивидуумы относительно того, как в среднем движется большинство генов. 9ч$| являются составляющими векторами. Из этого уравнения мы видим, что любой фактор нормализации или преобразования отменяется. VLR находится в диапазоне [0, ∞), где ноль указывает на идеальную координацию. В противном случае VLR не имеет значимой шкалы [36]. Таким образом, мы не можем сравнить VLR одной пары с VLR другой пары (поэтому вместо этого мы использовали пропорциональность) [30, 44]. Однако в дифференциальной пропорциональности мы сравниваем VLR для одной и той же пары по группам [31].
Мы интерпретируем пропорциональность на основе clr как обозначение координации, которая следует общей тенденции данных. Другими словами, эти пропорциональные гены движутся вместе как индивидуумы относительно того, как в среднем движется большинство генов. 9ч$| являются составляющими векторами. Из этого уравнения мы видим, что любой фактор нормализации или преобразования отменяется. VLR находится в диапазоне [0, ∞), где ноль указывает на идеальную координацию. В противном случае VLR не имеет значимой шкалы [36]. Таким образом, мы не можем сравнить VLR одной пары с VLR другой пары (поэтому вместо этого мы использовали пропорциональность) [30, 44]. Однако в дифференциальной пропорциональности мы сравниваем VLR для одной и той же пары по группам [31].
Дифференциальный анализ пропорциональности предназначен для выявления изменений пропорциональности между группами [31], интерпретируемых как изменение стехиометрии генов. Функция propd проверяет события, в которых коэффициент пропорциональности (т. е. величина x / y ) отличается между экспериментальными группами. Это измеряется θ d , которое колеблется от 0 до 1, где ноль указывает на максимальное различие между группами. Как и выше, пользователи могут переставлять FDR и строить сеть, но также могут вычислить точное значение P из θ d с помощью функции updateF [31] с дополнительным применением весов точности limma::voom [ 51] и F - статистическая модерация [52]. Точные веса устраняют зависимость средней дисперсии, влияющую на результаты при малом числе повторов, а модерируемая статистика помогает избежать ложноположительных результатов в случае нескольких повторов. При проверке значимости нескольких пар логарифмических отношений абсолютно необходимо исправить P -значение для многократного тестирования. Кроме того, эта функция реализует процедуру обработки нуля, основанную на преобразовании Бокса-Кокса, где α = 0,5, по-видимому, хорошо работает при моделировании (см.
е. величина x / y ) отличается между экспериментальными группами. Это измеряется θ d , которое колеблется от 0 до 1, где ноль указывает на максимальное различие между группами. Как и выше, пользователи могут переставлять FDR и строить сеть, но также могут вычислить точное значение P из θ d с помощью функции updateF [31] с дополнительным применением весов точности limma::voom [ 51] и F - статистическая модерация [52]. Точные веса устраняют зависимость средней дисперсии, влияющую на результаты при малом числе повторов, а модерируемая статистика помогает избежать ложноположительных результатов в случае нескольких повторов. При проверке значимости нескольких пар логарифмических отношений абсолютно необходимо исправить P -значение для многократного тестирования. Кроме того, эта функция реализует процедуру обработки нуля, основанную на преобразовании Бокса-Кокса, где α = 0,5, по-видимому, хорошо работает при моделировании (см. Дополнительный анализ S2). На рис. 4 показаны значимые дифференциально-пропорциональные пары, содержащие NFκB в логарифмическом отношении. Большинство этих сопутствующих генов также были названы ALDEx2 (относительно) дифференциально распространенными.
Дополнительный анализ S2). На рис. 4 показаны значимые дифференциально-пропорциональные пары, содержащие NFκB в логарифмическом отношении. Большинство этих сопутствующих генов также были названы ALDEx2 (относительно) дифференциально распространенными.
Рисунок 4:
Открыть в новой вкладкеСкачать слайд
График в параллельных координатах логарифмического соотношения (ось Y) значимых дифференциально пропорциональных пар, которые содержат NFκB в логарифмическом отношении (ось X). Каждая строка представляет один образец, окрашенный по группам. Пары генов слева от оси X имеют большие различия в средних логарифмических отношениях между группами (т. е. меньшее значение θ d значений). На этом графике показаны только пары, для которых образцы, стимулированные LPS, имеют разные средние значения логарифмического отношения от контроля (с порядком числителя и знаменателя, выбранным таким образом, чтобы среднее значение LPS всегда было больше, чем среднее значение в контроле). Неудивительно, что многие из этих значимых пар содержат одни и те же гены, обнаруженные дифференциальным анализом численности. Действительно, дифференциальный анализ пропорциональности можно рассматривать как дифференциальный анализ изобилия всех попарных логарифмических соотношений. Хотя пары справа от оси X по-прежнему имеют большие различия в логарифмическом отношении численности в среднем, некоторые моменты времени отклоняются от тенденции. Действительно, на этом рисунке случайно показан процесс, зависящий от времени, который мы могли бы протестировать специально с моделями, представленными в подразделе «Сложный план исследования»9.0003
Неудивительно, что многие из этих значимых пар содержат одни и те же гены, обнаруженные дифференциальным анализом численности. Действительно, дифференциальный анализ пропорциональности можно рассматривать как дифференциальный анализ изобилия всех попарных логарифмических соотношений. Хотя пары справа от оси X по-прежнему имеют большие различия в логарифмическом отношении численности в среднем, некоторые моменты времени отклоняются от тенденции. Действительно, на этом рисунке случайно показан процесс, зависящий от времени, который мы могли бы протестировать специально с моделями, представленными в подразделе «Сложный план исследования»9.0003
# propd ожидает:
# 'counts': матрица данных со строками в качестве выборок
# 'group': метки классов
Group = RNASEQ.Annot $ Обработка)
#Рассчитайте точное значение p-значения
PD <-Updatef (PD)
GetResults (PD)
Advanced Apprance
Комплексное исследование.
 наш конвейер для анализа данных, как если бы образцы принадлежали к 1 из 2 групп. Этот конвейер также может работать со сложными планами исследований с несколькими ковариатами. Для ALDEx2 мы можем предоставить объект R model.matrix, чтобы найти ожидание линейной модели (вместо т -тест). С другой стороны, пропорциональность рассчитывается для всех образцов, независимо от метки класса, и поэтому не требует новой процедуры. Дифференциальная пропорциональность измеряет разницу в логарифмическом отношении численности между двумя группами. По замыслу это эффективная реализация 2-группового дисперсионного анализа, выраженного формулой |${}[\log (\mathbf {x}_g) - \log (\mathbf {x}_h)] \sim \textrm{ group}$|, для всех комбинаций функций g и h . Таким образом, мы можем расширить дифференциальную пропорциональность, моделируя каждый парный результат логарифмического отношения как функцию любой модели model.matrix. Это может стать вычислительно обременительным для многомерных данных.
наш конвейер для анализа данных, как если бы образцы принадлежали к 1 из 2 групп. Этот конвейер также может работать со сложными планами исследований с несколькими ковариатами. Для ALDEx2 мы можем предоставить объект R model.matrix, чтобы найти ожидание линейной модели (вместо т -тест). С другой стороны, пропорциональность рассчитывается для всех образцов, независимо от метки класса, и поэтому не требует новой процедуры. Дифференциальная пропорциональность измеряет разницу в логарифмическом отношении численности между двумя группами. По замыслу это эффективная реализация 2-группового дисперсионного анализа, выраженного формулой |${}[\log (\mathbf {x}_g) - \log (\mathbf {x}_h)] \sim \textrm{ group}$|, для всех комбинаций функций g и h . Таким образом, мы можем расширить дифференциальную пропорциональность, моделируя каждый парный результат логарифмического отношения как функцию любой модели model.matrix. Это может стать вычислительно обременительным для многомерных данных.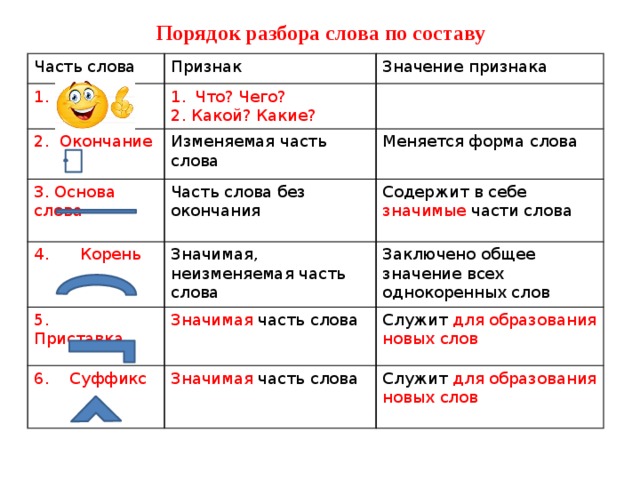 При проверке значимости нескольких пар логарифмических отношений абсолютно необходимо исправить P - значение для многократного тестирования, например, с использованием функции p.adjust в R.
При проверке значимости нескольких пар логарифмических отношений абсолютно необходимо исправить P - значение для многократного тестирования, например, с использованием функции p.adjust в R.Вертикальная интеграция данных
Мы предполагаем 2 общие стратегии для вертикальной интеграции композиционных данных. Во-первых, стратегия «соединения строк» рассматривает другие данные -omics как дополнительные образцы и моделирует источник -omics как ковариант. Это требует, чтобы все источники -omics сопоставлялись с одними и теми же функциями. Для данных RNA-Seq и MS, используемых здесь, оба количественно определяют относительное количество генных продуктов. Это позволяет нам использовать ALDEx2, чтобы найти признаки, в которых количество мРНК изменяется больше, чем содержание белка, относительно общего эталона (и наоборот). Аналогичным образом, мы можем использовать анализ пропорциональности, чтобы найти пары признаков, где гены и белки имеют скоординированную экспрессию в ответ на LPS. Наконец, мы можем использовать дифференциальный анализ пропорциональности, чтобы найти пары признаков со стехиометрическими различиями между парой генов и соответствующей парой белков. На рис. 5 показаны некоторые примеры дифференциально пропорциональных пар.
Наконец, мы можем использовать дифференциальный анализ пропорциональности, чтобы найти пары признаков со стехиометрическими различиями между парой генов и соответствующей парой белков. На рис. 5 показаны некоторые примеры дифференциально пропорциональных пар.
Рисунок 5:
Открыть в новой вкладкеСкачать слайд
Обилие мРНК по сравнению с обилием вновь синтезированного белка после стимуляции ЛПС, иллюстрирующее вертикальную интеграцию мультиомных данных в композиционной структуре. На левом поле мы показываем логарифмическое содержание 3 генов (MNDAL, SERPINB2 и PTGS2), измеренное с помощью RNA-Seq (оранжевый) и масс-спектрометрии (синий). Для композиционных данных эти изобилия не имеют смысла сами по себе, потому что ограниченная сумма накладывает «предвзятость закрытия». На верхнем поле мы показываем логарифмическое количество двух ссылок: RPL30 (выбрано, потому что его количество пропорционально среднему геометрическому значений образцов) и NFκB (выбрано на основе гипотезы). В середине мы показываем изобилие логарифмического отношения функции левого поля, деленное на ссылку верхнего поля (эквивалент левого поля минус верхнее поле в логарифмическом пространстве). Один только MNDAL, по-видимому, существует больше как мРНК, чем как белок, что остается верным при сравнении с обоими ссылками. Это говорит о том, что MNDAL транслируется с меньшей эффективностью, чем RPL30 и NKkB. С другой стороны, один только SERPINB2, по-видимому, в среднем существует в виде мРНК и белка; однако на самом деле он существует больше как белок, чем мРНК, если сравнивать с обоими ссылками. Это говорит о том, что MNDAL переводится с большей эффективностью, чем RPL30 и NKκB. Сам по себе PTGS2, по-видимому, существует больше в виде мРНК, чем белка, но это различие менее очевидно при сравнении с обоими эталонами. Это говорит о том, что PTGS2 транслируется с такой же эффективностью, как RPL30 и NKkB. Выбрав общий эталон между двумя наборами мультиомных данных, мы можем выполнить анализ вертикально интегрированных данных без необходимости нормализации.
В середине мы показываем изобилие логарифмического отношения функции левого поля, деленное на ссылку верхнего поля (эквивалент левого поля минус верхнее поле в логарифмическом пространстве). Один только MNDAL, по-видимому, существует больше как мРНК, чем как белок, что остается верным при сравнении с обоими ссылками. Это говорит о том, что MNDAL транслируется с меньшей эффективностью, чем RPL30 и NKkB. С другой стороны, один только SERPINB2, по-видимому, в среднем существует в виде мРНК и белка; однако на самом деле он существует больше как белок, чем мРНК, если сравнивать с обоими ссылками. Это говорит о том, что MNDAL переводится с большей эффективностью, чем RPL30 и NKκB. Сам по себе PTGS2, по-видимому, существует больше в виде мРНК, чем белка, но это различие менее очевидно при сравнении с обоими эталонами. Это говорит о том, что PTGS2 транслируется с такой же эффективностью, как RPL30 и NKkB. Выбрав общий эталон между двумя наборами мультиомных данных, мы можем выполнить анализ вертикально интегрированных данных без необходимости нормализации.
# Получить только обработанные ЛПС клетки0003
pro <- masshl.no0[masshl.annot$Treatment == “LPS'',]
# Объединить как единую матрицу («РНК», 14), rep («Белок», 14))
# Запустить анализ propd
pd.ms <- propd(merge, group)
Во-вторых, стратегия «объединения столбцов». рассматривает другие омиксные данные - как дополнительные признаки.Эта стратегия более сложна, поскольку требует, чтобы каждый -омический источник имел свою собственную ссылку.На практике мы должны выполнять дифференциальный анализ численности по каждому - источник omics самостоятельно. Для анализа пропорциональности и дифференциальной пропорциональности нам потребуется преобразовать логарифмическое отношение каждого источника - omics независимо, а затем объединить их в столбцы с помощью cbind. Здесь любая пропорциональность, возникающая между функциями из разных источников, будет относиться к двум ссылкам и должна интерпретироваться соответствующим образом.
Горизонтальная интеграция данных
Термин «мегаанализ» описывает единый анализ образцов, собранных в ходе нескольких исследований [53]. Пакетные эффекты представляют собой серьезное препятствие для мега-анализа. Здесь мы рассматриваем 2 типа пакетных эффектов. Первый пропорционально влияет на все гены в образце (например, из-за различий в глубине секвенирования). Преобразование логарифмического отношения автоматически удалит этот пакетный эффект. Второй влияет только на некоторые гены в образце (например, из-за различий в протоколах истощения РНК). Это требует явного изменения поврежденных функций. При необходимости можно применить стандартные инструменты пакетной коррекции, обычно применяемые к нормализованным данным, вместо этого к преобразованным данным (см. модерируемую лог-ссылку sva в [54]).
Кластеризация и классификация
В большинстве показателей расстояния отсутствует субкомпозиционное доминирование, а это означает, что можно уменьшить расстояние между выборками путем добавления измерений [16]. При кластеризации композиций методы, основанные на расстоянии, такие как иерархическая кластеризация, также не имеют субкомпозиционного доминирования [55]. Вместо этого следует использовать евклидово расстояние clr-преобразованных композиций (называемое расстоянием Эйчисона) [55]. Другие статистические методы, используемые для кластеризации, такие как анализ основных компонентов и t-распределенное стохастическое встраивание соседей (t-SNE), также вычисляют расстояние и также должны преобразовываться clr перед анализом. При кластеризации компонентов можно использовать метрику пропорциональности ϕ s в качестве меры несходства [30]. Метрика пропорциональности ϕ s , как и метрика пропорциональности ρ p , определена для данных, преобразованных в clr. Если среднее геометрическое резко меняется в выборках, некоторые пропорциональные пары могут не быть пропорциональными в абсолютном смысле. Мы отсылаем читателя к подразделу «Анализ пропорциональности с помощью propr» для дальнейшего разъяснения.
При кластеризации композиций методы, основанные на расстоянии, такие как иерархическая кластеризация, также не имеют субкомпозиционного доминирования [55]. Вместо этого следует использовать евклидово расстояние clr-преобразованных композиций (называемое расстоянием Эйчисона) [55]. Другие статистические методы, используемые для кластеризации, такие как анализ основных компонентов и t-распределенное стохастическое встраивание соседей (t-SNE), также вычисляют расстояние и также должны преобразовываться clr перед анализом. При кластеризации компонентов можно использовать метрику пропорциональности ϕ s в качестве меры несходства [30]. Метрика пропорциональности ϕ s , как и метрика пропорциональности ρ p , определена для данных, преобразованных в clr. Если среднее геометрическое резко меняется в выборках, некоторые пропорциональные пары могут не быть пропорциональными в абсолютном смысле. Мы отсылаем читателя к подразделу «Анализ пропорциональности с помощью propr» для дальнейшего разъяснения.
Как лучше всего классифицировать композиционные данные, остается открытым вопросом, но ilr-преобразование данных перед обучением модели придаст данным благоприятные свойства, как это делается для линейного дискриминантного анализа [56]. В качестве альтернативы можно обучать модели на самих логарифмических отношениях, хотя это может не масштабироваться для многомерных данных. В последнее время балансы используются для выбора и классификации признаков [57,58], где они обеспечивают как точность, так и интерпретируемость [59].].
Избранные темы
Смещение закрытия и неявная ссылка
Данные подсчета NGS измеряют относительную численность из-за произвольного ограничения, налагаемого клеткой, окружающей средой и секвенсором. Это иногда называют «ограничением постоянной суммы», потому что сумма относительных содержаний должна равняться константе. Все, что вводит ограничение на постоянную сумму, является своего рода «замыканием»; все замыкания необратимо делают набор данных относительным (то есть «закрытым»). Можно думать о клетке (в случае RNA-Seq) или окружающей среде (в случае метагеномики) как о естественных замыканиях, а о секвенаторах — как о технических замыканиях.
Можно думать о клетке (в случае RNA-Seq) или окружающей среде (в случае метагеномики) как о естественных замыканиях, а о секвенаторах — как о технических замыканиях.
Нормализация общего размера библиотеки, как и TPM, вообще не является нормализацией: на самом деле это еще одно замыкание, накладывающее ограничение на постоянную сумму транскриптов на миллион. TPM не преобразуют закрытые данные секвенирования в «открытые» единицы, такие как концентрация. Анализ TPM, как если бы они были концентрациями, теоретически ошибочен и может существенно повлиять на моделирование клеточных процессов. Наш собственный анализ показывает, что в Jovanovic et al. [27], скорость трансляции мРНК могла быть систематически завышена из-за композиционной предвзятости. В дополнительном анализе S1 мы показываем, что в самый последний момент времени ошибка по сравнению с нормализованными данными составляет ~ 13% в контрольных условиях и достигает 35% в образцах, стимулированных ЛПС. Это смещение связано с операцией замыкания: если аналитик не выбирает эталон, оценки должны интерпретироваться с учетом неизвестного и неизмеримого «смещения замыкания». Поскольку величина этого смещения закрытия может быть большой для образцов, которые широко варьируются с точки зрения способности к синтезу нуклеотидов, всегда следует использовать эталон при моделировании одномерных характеристик композиционных данных. Если ссылка не выбрана, то смещение закрытия действует как «неявная ссылка», что делает интерпретацию невозможной.
Поскольку величина этого смещения закрытия может быть большой для образцов, которые широко варьируются с точки зрения способности к синтезу нуклеотидов, всегда следует использовать эталон при моделировании одномерных характеристик композиционных данных. Если ссылка не выбрана, то смещение закрытия действует как «неявная ссылка», что делает интерпретацию невозможной.
Композиции подсчета и низкая неточность подсчета
Данные закрытого подсчета отличаются от идеализированных композиционных данных, потому что аддитивные вариации влияют на небольшие подсчеты больше, чем на большие [30]. Таким образом, разница между 1 и 2 счетами не совпадает с разницей между 1000 и 2000 счетами. Более того, эксперименты NGS часто имеют гораздо больше характеристик, чем выборки, что приводит к серьезной недооценке технической дисперсии; действительно, техническая дисперсия может быть намного больше, чем биологическая дисперсия на границе малого счета [29]. ]. Признаки «нулевого счета» — это те, которые наблюдаются как ненулевые значения в ≥1 выборке и, таким образом, ожидается, что они будут наблюдаться на границе или рядом с ней в других выборках. Хотя это и не интуитивно понятно, распределение относительных нулевых значений счета довольно велико и охватывает многие порядки величины [60]. Кроме того, ожидаемое значение нулевого признака счета должно быть больше нуля, поскольку значение больше нуля наблюдалось в ≥1 образце.
]. Признаки «нулевого счета» — это те, которые наблюдаются как ненулевые значения в ≥1 выборке и, таким образом, ожидается, что они будут наблюдаться на границе или рядом с ней в других выборках. Хотя это и не интуитивно понятно, распределение относительных нулевых значений счета довольно велико и охватывает многие порядки величины [60]. Кроме того, ожидаемое значение нулевого признака счета должно быть больше нуля, поскольку значение больше нуля наблюдалось в ≥1 образце.
Как упоминалось выше, нулевые значения счетчика могут быть изменены для получения точечной оценки их ожидаемого значения, но это приводит к недооценке их истинной дисперсии, поскольку мы оцениваем ожидаемое значение признака. В подходе, реализованном в функции aldex.clr, используемой функциями ALDEx2::aldex.ttest, ALDEx2::aldex.effect и propr::aldex2propr, распределение нулевых значений счетчика определяется путем выборки из распределения Дирихле (т.е. , многомерное обобщение β-распределения). Другой способ представить распределение Дирихле — это многомерная выборка Пуассона с ограничением постоянной суммы. Распределение относительной численности вблизи границы низкого учета может быть удивительно широким, как по оценкам выборки из распределения Дирихле, так и по реальным данным [60]. Делая выборку из распределения Дирихле, мы получаем набор многомерных векторов вероятности, каждый из которых с такой же вероятностью наблюдался из исходных данных, как и тот, который фактически наблюдался из секвенированной выборки. Исходя из этого, ALDEx2 и propr могут объяснить небольшую техническую неточность (которая может быть намного больше, чем биологическая вариация), сообщая ожидаемые значения тестовой статистики вместо точечной оценки [29].].
Распределение относительной численности вблизи границы низкого учета может быть удивительно широким, как по оценкам выборки из распределения Дирихле, так и по реальным данным [60]. Делая выборку из распределения Дирихле, мы получаем набор многомерных векторов вероятности, каждый из которых с такой же вероятностью наблюдался из исходных данных, как и тот, который фактически наблюдался из секвенированной выборки. Исходя из этого, ALDEx2 и propr могут объяснить небольшую техническую неточность (которая может быть намного больше, чем биологическая вариация), сообщая ожидаемые значения тестовой статистики вместо точечной оценки [29].].
Всплеск «нормализации логарифмического отношения»
Преобразования не являются нормализацией, поскольку они не претендуют на преобразование данных в абсолютном выражении. Однако, если бы кто-то выбрал набор эталонов с априорно известным фиксированным содержанием во всех образцах, можно было бы использовать этот «идеальный эталон» для нормализации данных (то, что мы называем «нормализация логарифмического отношения» [14]). Одним из таких вариантов может быть использование вводимых контролей, состоящих из нескольких синтетических нуклеотидных последовательностей с известным абсолютным содержанием. Для RNA-Seq набор вставок Консорциума по контролю внешней РНК (ERCC) состоит из 92 полиаденилированных РНК-транскриптов с различной длиной (250–2000 нт) и содержанием гуанин-цитозина (5–51%) с 10 6 -кратным диапазоном численности [61]. Набор для добавления добавляют к стандартному количеству очищенной РНК в эквимолярных концентрациях; затем как добавленный, так и целевой транскрипты обрабатываются вместе для создания библиотеки кДНК. Поскольку 23 транскрипта ERCC имеют одинаковую абсолютную распространенность, можно использовать их среднее геометрическое в качестве эталона для пересчета данных в абсолютном выражении. Точно так же можно добавить известное количество бактериальных клеток или синтетических плазмид, чтобы стандартизировать количество метагеномных образцов, амплифицированных с помощью ПЦР [62, 63].
Одним из таких вариантов может быть использование вводимых контролей, состоящих из нескольких синтетических нуклеотидных последовательностей с известным абсолютным содержанием. Для RNA-Seq набор вставок Консорциума по контролю внешней РНК (ERCC) состоит из 92 полиаденилированных РНК-транскриптов с различной длиной (250–2000 нт) и содержанием гуанин-цитозина (5–51%) с 10 6 -кратным диапазоном численности [61]. Набор для добавления добавляют к стандартному количеству очищенной РНК в эквимолярных концентрациях; затем как добавленный, так и целевой транскрипты обрабатываются вместе для создания библиотеки кДНК. Поскольку 23 транскрипта ERCC имеют одинаковую абсолютную распространенность, можно использовать их среднее геометрическое в качестве эталона для пересчета данных в абсолютном выражении. Точно так же можно добавить известное количество бактериальных клеток или синтетических плазмид, чтобы стандартизировать количество метагеномных образцов, амплифицированных с помощью ПЦР [62, 63].
Однако в основе использования всплесков для нормализации лежат два важных допущения. Во-первых, предполагается, что последовательности-вставки и последовательности-мишени имеют одинаковую эффективность захвата преобразования РНК, поскольку на них одинаково влияют технические предубеждения при создании библиотеки кДНК. Во-вторых, предполагается, что всплески откалиброваны по количеству молекул РНК на клетку. Другими словами, предполагается, что на молекулу РНК и добавляется такое количество вставки, что каждая клетка продуцирует одинаковое количество молекул РНК. Последнее представляет собой особую проблему для массового RNA-Seq из-за технической сложности добавления соответствующего количества вставки на уровне клеточной популяции [64]. Однако даже при контроле технических вариаций клетки могут продуцировать меньше общей РНК в 1 из экспериментальных групп [10] или с течением времени [65]. В этом случае стандартизация всплеска к общему количеству вводимой РНК сделает это предположение недействительным.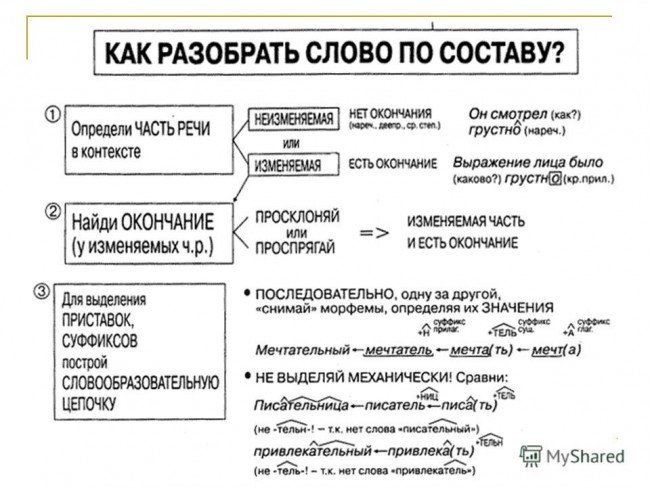 Без стандартизации прибавления к общему количеству клеток невозможно восстановить абсолютную численность (т. е. в единицах транскриптов на клетку) [66]. Даже если бы можно было стандартизировать всплески для общего числа клеток, интерпретация может быть затруднена, если клетки в одной партии производят различное количество общей РНК.
Без стандартизации прибавления к общему количеству клеток невозможно восстановить абсолютную численность (т. е. в единицах транскриптов на клетку) [66]. Даже если бы можно было стандартизировать всплески для общего числа клеток, интерпретация может быть затруднена, если клетки в одной партии производят различное количество общей РНК.
Помимо всплесков ERCC, было предложено несколько других всплесков. Для исследований RNA-Seq примеры вставок включают блестки [67, 68], контрольные плазмидные геномы с вставками [69] и изоформ-специфические варианты вставки РНК [70]. Для исследований метагеномики примерами всплесков являются экзогенные бактерии [62] и блестки [71]. В задачи данного полевого руководства не входит сравнение и противопоставление всех различных всплесков. Однако мы должны подчеркнуть, что если всплески откалиброваны по общему весу вводимой РНК, они не приводят автоматически данные к абсолютной численности. Причина этого логически вытекает из того, как работают вставки: когда вставки добавляются в фиксированной пропорции к произвольной массе РНК, секвенирование будет возвращать количество в той же фиксированной пропорции. Таким образом, всплески говорят нам только о количестве секвенированной РНК. Однако термин «абсолютное содержание» относится к количеству РНК, присутствующей в биологическом образце (например, в единицах транскриптов на клетку для РНК-Seq или бактерий на литр для метагеномики). Следовательно, всплески нормализуются до абсолютного содержания тогда и только тогда, когда количество секвенированной РНК равно количеству РНК, присутствующей в биологическом образце. Даже если разница между абсолютной РНК и исходной РНК, которую мы называем δ, пропорциональна, эта δ должна быть одинаковой для всех образцов. В противном случае δ становится еще одним смещением замыкания, которое может привести к систематическим ошибкам. В этом случае «нормализация» всплеска вызывает ту же проблему, что и «нормализация» TPM: аналитик преобразовал свои старые композиции в новые композиции, ошибочно полагая, что новые композиции представляют собой абсолютные концентрации. Прежде чем использовать нормализацию всплесков, аналитик должен критически оценить свой протокол, чтобы оценить, могут ли они с уверенностью предположить, что δ фиксировано для всех образцов.
Таким образом, всплески говорят нам только о количестве секвенированной РНК. Однако термин «абсолютное содержание» относится к количеству РНК, присутствующей в биологическом образце (например, в единицах транскриптов на клетку для РНК-Seq или бактерий на литр для метагеномики). Следовательно, всплески нормализуются до абсолютного содержания тогда и только тогда, когда количество секвенированной РНК равно количеству РНК, присутствующей в биологическом образце. Даже если разница между абсолютной РНК и исходной РНК, которую мы называем δ, пропорциональна, эта δ должна быть одинаковой для всех образцов. В противном случае δ становится еще одним смещением замыкания, которое может привести к систематическим ошибкам. В этом случае «нормализация» всплеска вызывает ту же проблему, что и «нормализация» TPM: аналитик преобразовал свои старые композиции в новые композиции, ошибочно полагая, что новые композиции представляют собой абсолютные концентрации. Прежде чем использовать нормализацию всплесков, аналитик должен критически оценить свой протокол, чтобы оценить, могут ли они с уверенностью предположить, что δ фиксировано для всех образцов. С другой стороны, на преобразование по отношению к внутренней ссылке не влияют глобальные различия в δ.
С другой стороны, на преобразование по отношению к внутренней ссылке не влияют глобальные различия в δ.
Секвенирование РНК одиночных клеток
Секвенирование РНК одиночных клеток (scRNA-Seq) похоже на групповое RNA-Seq, за исключением того, что РНК отдельных клеток захватывается и штрих-кодируется отдельно перед созданием библиотеки кДНК [72]. Этот этап захвата РНК включает неисчерпывающую выборку общей РНК, которая действует как еще одна операция закрытия, чтобы сделать данные относительными. Затем секвенсор повторно закроет уже закрытые данные. Интересно, что если бы библиотеки последовательностей затем выражали в TPM, делитель на миллион действовал бы как еще одно замыкание данных. По этим причинам scRNA-Seq напоминает другие данные подсчета NGS в том смысле, что каждый образец представляет собой композицию относительных частей. Как и другие данные подсчета NGS, невозможно оценить абсолютное количество РНК без эталонного пика на клетку.
Анализ scRNA-Seq описывается как более сложный, чем массовый анализ RNA-Seq по двум причинам. Во-первых, размеры библиотеки scRNA-Seq больше различаются между образцами [73]. Это связано с различиями в эффективности захвата при экстракции РНК, глубине секвенирования и так называемых «двойных» событиях, когда захватываются сразу 2 клетки [73]. Чтобы устранить эти различия в размере библиотеки, данные нормализуются с помощью нормализации эффективного размера библиотеки или эталонной нормализации (через набор расшифровок служебных или всплесковых расшифровок). Нормализация эффективного размера библиотеки предполагает, что большинство генов не изменены; это предположение особенно проблематично для данных scRNA-Seq, поскольку в экспериментах с одиночными клетками изучаются гетерогенные клеточные популяции [74]. Нормализация ссылок также имеет ограничения. Гены домашнего хозяйства могут не иметь последовательной экспрессии на уровне одной клетки из-за разрыва транскрипции или тканевой гетерогенности [74]. Между тем, вставки scRNA-Seq подразумевают те же предположения, что и массовая RNA-Seq: вставки и последовательности-мишени имеют одинаковую эффективность захвата преобразования РНК и что вставки откалиброваны по количеству молекул РНК на клетку. . Второе допущение проблематично для scRNA-Seq, поскольку подразумевает, что эффективность захвата экстракции РНК оказывает одинаковое влияние на все клетки [74]. Поскольку вставки добавляются в буфер для лизиса, нормализация всплесков может показать только то, сколько РНК было захвачено из клетки, а не количество РНК, присутствующее в клетке: таким образом, всплески не могут нормализовать различия в лизисе клеток. эффективности (что является обычным явлением и является важной причиной «отсева») [75]. С другой стороны, на трансформацию по отношению к внутреннему эталону не влияют глобальные различия в эффективности лизиса клеток. Это аналогично обсуждению δ из предыдущего пункта.
Между тем, вставки scRNA-Seq подразумевают те же предположения, что и массовая RNA-Seq: вставки и последовательности-мишени имеют одинаковую эффективность захвата преобразования РНК и что вставки откалиброваны по количеству молекул РНК на клетку. . Второе допущение проблематично для scRNA-Seq, поскольку подразумевает, что эффективность захвата экстракции РНК оказывает одинаковое влияние на все клетки [74]. Поскольку вставки добавляются в буфер для лизиса, нормализация всплесков может показать только то, сколько РНК было захвачено из клетки, а не количество РНК, присутствующее в клетке: таким образом, всплески не могут нормализовать различия в лизисе клеток. эффективности (что является обычным явлением и является важной причиной «отсева») [75]. С другой стороны, на трансформацию по отношению к внутреннему эталону не влияют глобальные различия в эффективности лизиса клеток. Это аналогично обсуждению δ из предыдущего пункта.
Во-вторых, scRNA-Seq содержит много нулей. Хотя некоторые нули описываются как «биологические нули» (т. е. существенные нули) [76], большинство из них описываются как «нули выпадения». Для отсева нуль — это отсутствующее значение, которое возникает из-за того, что «молекулы мРНК не захватываются... в одинаковой пропорции» для всех клеток [72]. Согласно этому определению, нули отсева — это просто «нули счета», вызванные неполным отбором проб. Поскольку различия в эффективности лизиса клеток являются важной причиной отсева, всплески не могут решить проблему отсева [75]. Однако эти нули отсева являются на самом деле ничем не отличается от нулей недостаточной выборки, найденных в метагеномных данных (которые уже обрабатываются нашим конвейером [29].]). Однако, если аналитик желает вменить нули, существуют методы вменения, разработанные специально для композиционных данных [77,78].
е. существенные нули) [76], большинство из них описываются как «нули выпадения». Для отсева нуль — это отсутствующее значение, которое возникает из-за того, что «молекулы мРНК не захватываются... в одинаковой пропорции» для всех клеток [72]. Согласно этому определению, нули отсева — это просто «нули счета», вызванные неполным отбором проб. Поскольку различия в эффективности лизиса клеток являются важной причиной отсева, всплески не могут решить проблему отсева [75]. Однако эти нули отсева являются на самом деле ничем не отличается от нулей недостаточной выборки, найденных в метагеномных данных (которые уже обрабатываются нашим конвейером [29].]). Однако, если аналитик желает вменить нули, существуют методы вменения, разработанные специально для композиционных данных [77,78].
Обсуждение
CoDA предоставляет концептуальную основу для изучения относительных данных. В этой статье мы представляем набор программных инструментов, предназначенных для данных подсчета NGS, которые вместе образуют конвейер, который объединяет анализ всех композиционных данных, включая RNA-Seq, метагеномные данные, данные об отдельных клетках и спектрометрические пики. В отличие от существующих конвейеров, наш не стремится нормализовать данные, чтобы восстановить абсолютное изобилие. Вместо этого он преобразует данные относительно эталона, позволяя аналитику изучать любой относительный набор данных, не прибегая к часто непроверяемым предположениям, лежащим в основе нормализации данных NGS.
В отличие от существующих конвейеров, наш не стремится нормализовать данные, чтобы восстановить абсолютное изобилие. Вместо этого он преобразует данные относительно эталона, позволяя аналитику изучать любой относительный набор данных, не прибегая к часто непроверяемым предположениям, лежащим в основе нормализации данных NGS.
Платформа CoDA развилась независимо от многих альтернативных методов, применяемых в настоящее время к данным NGS. Интересно, что, хотя они и не были специально адаптированы для композиционных данных, самые строгие из методов NGS сошлись на аналогичных решениях для обработки композиционной погрешности. Они основаны на эффективной нормализации размера библиотеки (и смещениях), которая использует (псевдо-счетные) логарифмически преобразованные данные аналогично преобразованиям логарифмического отношения. В CoDA такие преобразования явно выводятся для учета ограниченного характера данных. С этой точки зрения явные ссылки и попарные логарифмические соотношения применимы к более широкому кругу экспериментов, включая менее контролируемые исследования, в которых эффективная нормализация размера библиотеки может не работать. Анализ композиций подсчета, особенно обработка неточности при низком подсчете, в настоящее время достиг состояния зрелости, которое позволяет проводить анализ NGS без какой-либо потери формальной строгости.
Анализ композиций подсчета, особенно обработка неточности при низком подсчете, в настоящее время достиг состояния зрелости, которое позволяет проводить анализ NGS без какой-либо потери формальной строгости.
Важным аспектом CoDA является то, что он лучше количественно определяет координацию между функциями, чем корреляцию, последняя из которых часто оказывается ложной, когда игнорируется композиционное ограничение. Между тем, применение дифференциального анализа численности по отношению к эталону остается в силе даже в самых разнообразных условиях. Для кластеризации и классификации расстояние Эйтчисона, полностью основанное на отношении, обеспечивает превосходное межвыборочное расстояние, которое до сих пор недооценивается в современных приложениях. И последнее, но не менее важное: CoDA открывает новые перспективы в отношении интеграции больших наборов мультиомных данных, где явные ссылки могут сыграть важную роль в будущем.
Наличие исходного кода и требований
Название проекта: CoDa-Protocol
Домашняя страница проекта: http://doi.
org/10.5281/zenodo.32709542
9 90 независимых операционных систем
Язык программирования: R
Прочие требования: Пакеты R zCompositions, ALDEx2, propr, patchwork, ggplot2, Knitr и Plyr
Лицензия: GPLv3
 org/10.5281/zenodo.3270954
org/10.5281/zenodo.3270954Наличие вспомогательных данных и материалов
Все данные и сценарии находятся в открытом доступе на http://doi.org/10.5281/zenodo.3270954 [79].
Дополнительные файлы
Дополнительная информация : Дополнительные методы и результаты доступны в дополнительном файле, связанном с этой статьей.
Дополнительный анализ S1: supp-1.pdf
Дополнительный анализ S2: suppZero.pdf
Сокращения
alr: аддитивный логарифмический коэффициент; ANOVA: дисперсионный анализ; кДНК: комплементарная ДНК; clr: центрированное логарифмическое отношение; CoDa: композиционные данные; CoDA: композиционный анализ данных; CRAN: комплексная сеть R-архивов; DA: дифференциальная численность; ERCC: Консорциум по внешнему контролю РНК; FDR: частота ложных открытий; ilr: изометрический логарифмический коэффициент; iqlr: межквартильный логарифмический коэффициент; ЛПС: липополисахарид; malr: мультиаддитивный логарифмический коэффициент; мРНК: информационная РНК; МС: масс-спектрометрия; NFκB: ядерный фактор κB; NGS: секвенирование следующего поколения; OTU: операционная таксономическая единица; rclr: надежный центрированный логарифмический коэффициент; RNA-Seq: секвенирование РНК; scRNA-Seq: секвенирование одноклеточной РНК; TPM: число транскриптов на миллион; VLR: логарифмическая дисперсия.
Конкурирующие интересы
Авторы заявляют, что у них нет конкурирующих интересов.
Вклад авторов
T.P.Q. набросал и составил полевой путеводитель. Т.П.К., И.Е., Г.Г. и М.Ф.Р. составлен раздел «Избранные темы». т.е. подготовлен дополнительный анализ S1. Т.П.К. подготовлен дополнительный анализ S2. C.N., M.F.R. и T.M.C. курировал проект. Все авторы пересмотрели и одобрили окончательный вариант рукописи.
БЛАГОДАРНОСТИ
T.P.Q. спасибо Ларри Крофту за полезные обсуждения.
ССЫЛКИ
1.
Metzker
ML
.
Технологии секвенирования - следующее поколение
.
Nat Rev Genet
.
2010
;
11
(
1
):
31
–
46
.
2.
Вули
JC
,
Годзик
А
,
Фридберг
я
.
A Учебник по метагеномике
.
PLoS Comput Biol
.
2010
;
6
(
2
):
e1000667
.
3.
Башиардес
S
,
Зильберман-Шапира
G
,
3
03 Elinav
3 .

Использование метатранскриптомики в исследованиях микробиома
.
Биоинформ Биол Инсайтс
.
2016
;
10
:
19
–
25
.
4.
Парк
ПД
.
ChIP-Seq: преимущества и проблемы развивающейся технологии
.
Nat Rev Genet
.
2009
;
10
(
10
):
669
–
80
.
5.
Головка
SR
,
Komori
HK
,
LaMere
4
SA. .
Создание библиотеки для секвенирования следующего поколения: обзоры и задачи
.
БиоТехники
.
2014
;
56
(
2
):
61
–
пассим
.
6.
Торсен
J
,
Брейнрод
А
,
Мортенсен
4
М. .Крупномасштабный бенчмаркинг выявляет ложные открытия и чувствительность преобразования подсчета в методах анализа данных ампликона гена 16S рРНК, используемых в исследованиях микробиома
.
Микробиом
.
2016
;
4
:
62
.
7.
Hawinkel
S
,
Mattiello
F
,
Bijnens
.et
.
Нарушенное обещание: методы дифференциальной численности микробиома не контролируют частоту ложных открытий
.
Бриф Биоинформ
.
2019
,
20
,
1
,
210
–
21
.
8.
Фернандес
AD
,
Рейд
JN
,
Маклейм
. .
Унификация анализа наборов данных высокопроизводительного секвенирования: характеристика РНК-секвенирования, секвенирования гена 16S рРНК и экспериментов по селективному росту с помощью анализа композиционных данных
.
Микробиом
.
2014
;
2
:
15
.
9.
ван ден Бугаарт
KG
,
Толосана-Дельгадо
R
.
Описательный анализ композиционных данных
. В:
Анализ композиционных данных с помощью R. Используйте R!
.
Берлин, Гейдельберг
:
Springer
;
2013
:
73
–
93
.
10.
Lovén
J
,
Orlando
DA
,
Sigova
AA
, et al. .
Повторный анализ глобальной экспрессии генов
.
Сотовый
.
2012
;
151
(
3
):
476
–
82
.
11.
Падован-Мерхар
O
,
Наир
GP
,
Биэш
AG
, 9023 и др. .
Отдельные клетки млекопитающих компенсируют различия в клеточном объеме и количестве копий ДНК с помощью независимых глобальных механизмов транскрипции
.
Molec Cell
.
2015
;
58
(
2
):
339
–
52
.
12.
Эйтчисон
J
.
Краткое руководство по анализу композиционных данных
. В:
2-й семинар по анализу композиционных данных, Жирона, Испания
.
2003
.
.
13.
Gloor
ГБ
,
Macklaim
JM
,
Pawlowsky-Glahn
V
, et al. .
Наборы данных микробиома являются составными: и это необязательно
.
Фронт Микробиол
.
2017
;
8
:
2224
.
14.
Куинн
TP
,
Эрб
I
,
Ричардсон
MF et al. .
Понимание данных секвенирования как композиций: обзор и обзор
.
Биоинформатика
.
2018
;
34
(
16
):
2870
–
8
.
15.
ван ден Бугаарт
KG
,
Толосана-Дельгадо
R
.
«композиции»: унифицированный пакет R для анализа композиционных данных
.
Comput Geosci
.
2008
;
34
(
4
):
320
–
38
.
16.
Aitchison
J
,
Barceló-Vidal
C
,
Martín-Fernández
JA
, et al. .
Логарифмический анализ и композиционное расстояние
.
Мат Геол
.
2000
;
32
(
3
):
271
–
5
.
17.
Пирсон
К
.
Математический вклад в теорию эволюции. III. Регрессия, наследственность и панмиксия
.
Philos Trans R Soc Lond A
.
1896
;
187
:
253
–
318
.
18.
Фильцмозер
P
,
Валчак
B
.
Что может пойти не так на этапе нормализации данных для идентификации биомаркеров?
.
Ж Хроматогр А
.
2014
;
1362
:
194
–
205
.
19.
Робинсон
МД
,
Ошлак
А
.
Метод нормализации масштабирования для анализа дифференциальной экспрессии данных секвенирования РНК
.
Геном Биол
.
2010
;
11
:
R25
.
20.
Андерс
S
,
Хубер
W
.
Анализ дифференциальной экспрессии для данных подсчета последовательностей
.
Геном Биол
.
2010
;
11
:
R106
.
21.
Mateu-Figueras
G
,
Pawlowsky-Glahn
V
,
Egozcue
JJ
.
Принцип работы по координатам
. В:
Павловски-Глан
V
,
Buccianti
A
, ред.
Композиционный анализ данных
.
Wiley
;
2011
:
29
–
42
.
22.
Гринакр
М
.
Выбор переменных в анализе композиционных данных с использованием попарных логарифмических соотношений
.
Math Geosci
.
2018
;
51
(
5
):
649
–
82
.
23.
Engström
PG
,
STEIJGER
T
,
SIPOS
B
, и др. .
Систематическая оценка программ выравнивания сплайсинга для данных секвенирования РНК
.
Естественные методы
.
2013
;
10
(
12
):
1185
–
91
.
24.
Fonseca
NA
,
Rung
J
,
Brazma
3 ,
3 al. .
Инструменты для картирования высокопроизводительных данных секвенирования
.
Биоинформатика
.
2012
;
28
(
24
):
3169
–
77
.
25.
Патро
Р
,
Дуггал
G
,
Любовь
МИ
и др. .
Лосось: быстрый и предусмотрительный количественный анализ экспрессии транскриптов с использованием двухфазного вывода
.
Естественные методы
.
2017
;
14
(
4
):
417
.
26.
Трапнелл
С
,
Уильямс
BA
,
Pertea
G
и др. .
Сборка транскриптов и количественный анализ с помощью RNA-Seq выявляют неаннотированные транскрипты и переключение изоформ во время дифференцировки клеток
.
Нат Биотехнолог
.
2010
;
28
(
5
):
511
–
5
.
27.
Йованович
M
,
Rooney
MS
,
Mertins
P
, и др. .
Динамическое профилирование жизненного цикла белков в ответ на патогены
.
Наука
.
2015
;
347
(
6226
):
1259038
.
28.
Палареа Альбаладехо
J
,
Фернандес
М
,
Антони
Дж
.
Пакет zCompositions — R для многомерного вменения данных с цензурой слева при композиционном подходе
. Хемометрика и интеллектуальные лабораторные системы
2015
:143(1):85-96.
29.
Fernandes
г. н.э.
,
Macklaim
JM
,
Linn
TG
, et al. .
ANOVA-подобный анализ дифференциальной экспрессии (ALDEx) для смешанной популяции RNA-Seq
.
PLoS One
.
2013
;
8
(
7
):
e67019
.
30.
Куинн
TP
,
Ричардсон
MF
,
Ловелл
D .propr: R-пакет для определения пропорционально обильных признаков с использованием композиционного анализа данных
.
Научный представитель
.
2017
;
7
(
1
):
16252
.
31.
Эрб
I
,
Куинн
T
,
Ловелл
9 02303 D . .Дифференциальная пропорциональность - свободный от нормализации подход к дифференциальной экспрессии генов. Материалы CoDaWork 2017, 7-й семинар по анализу композиционных данных
.
биоРксив
.
2017
, doi:
10.1101/134536
.
32.
Куинн
TP
,
Кроули
TM
,
Ричардсон
3MF
Сравнительный анализ инструментов анализа дифференциальной экспрессии для RNA-Seq: методы, основанные на нормализации, и методы, основанные на логарифмическом соотношении
.
Биоинформатика BMC
.
2018
;
19
:
274
.
33.
Skinnider
MA
,
Squair
JW
,
Foster
3
J 39003
Оценка показателей ассоциации для транскриптомики одиночных клеток
.
Естественные методы
.
2019
;
16
(
5
):
381
–
6
.
34.
Martín-Fernández
JA
,
Palarea-Albaladejo
J
,
OLEA
RA
.
Работа с нулями
. In:
Композиционный анализ данных
.
Wiley-Blackwell
;
2011
:
43
–
58
., doi:
10.1002/9781119976462.ch5
.
35.
Сильверман
JD
,
Рош
К
,
Мукерджи
3
9000 .Нет Все нули в данных счетчика последовательности одинаковы
.
биоРксив
.
2018
, doi:
10.1101/477794
.
36.
Эйтчисон
Дж
.
Статистический анализ композиционных данных
.
Лондон, Великобритания
:
Chapman & Hall
;
1986
.
37.
WU
JR
,
Macklaim
JM
,
Genge
BL
, et al. .
Нахождение центра: поправки на асимметрию в наборах данных высокопроизводительного секвенирования
.
архив
.
2017
:
1704.01841
.
38.
Martino
C
,
Morton
JT
,
Marotz
, et al. .Новый метод разреженной композиции выявляет микробные возмущения
.
mSystems
.
2019
;
4
(
1
):
e00016
–
19
.
39.
Egozcue
JJ
,
Pawlowsky-Glahn
V
,
Mateu-Figueras
G
, et al. .
Изометрические логарифмические преобразования для композиционного анализа данных
.
Мат Геол
.
2003
;
35
(
3
):
279
–
300
.
40.
Silverman
JD
,
Washburne
AD
,
Mukherjee
S
, и др. .
Филогенетическое преобразование улучшает анализ данных о составе микробиоты
.
eLife
.
2017
;
6
, doi:
10.7554/eLife.21887
.
41.
Уошберн
AD
,
Сильверман
JD
,
Лефф
3JW
.
Филогенетическая факторизация композиционных данных дает ассоциации на уровне родословных в наборах данных микробиома
.
PeerJ
.
2017
;
5
:
e2969
.
42.
Кумар
МС
,
Слуд
ЭВ
,
Окра
К
и др. .
Анализ и коррекция композиционной погрешности в разреженных данных подсчета секвенирования
.
BMC Genomics
.
2018
;
19
(
1
):
799
.
43.
Pålsson-McDermott
EM
,
О’Нил
LAJ
.
Передача сигнала липополисахаридным рецептором, Toll-подобный рецептор-4
.
Иммунология
.
2004
;
113
(
2
):
153
–
62
.
44.
Ловелл
D
,
Павловски-Глан
В
,
Эгоскью
JJ
и др. .
Пропорциональность: достойная альтернатива корреляции относительных данных
.
PLoS Comput Biol
.
2015
;
11
(
3
):
e1004075
.
45 .
Как мы должны измерить пропорциональность данных об относительной экспрессии генов?
.
Theor Biosci
.
2016
;
135
:
21
–
36
.
46.
Фридман
J
,
Альм
EJ
.
Вывод корреляционных сетей из данных геномного исследования
.
PLoS Comput Biol
.
2012
;
8
(
9
):
e1002687
.
47.
Kurtz
ZD
,
Мюллер
CL
,
Miraldi
3
.Разреженный и композиционно надежный вывод о микробных экологических сетях
.
PLoS Comput Biol
.
2015
;
11
(
5
):
e1004226
.
48.
Биан
Г
,
Глор
ГБ
,
Гонг
А А и др. .Микробиота кишечника здоровых пожилых китайцев аналогична микробиоте здоровых молодых людей
.
mSphere
.
2017
;
2
(
5
):
e00327
–
17
.
49.
Гринакр
М
.
Измерение субкомпозиционной несвязности
.
Math Geosci
.
2011
;
43
(
6
):
681
–
93
.
50.
Валах
J
,
Фильцмозер
P
,
Грон
3
Kал. .Надежная идентификация биомаркеров в задаче с двумя классами на основе попарных логарифмических соотношений
.
Хемометр Intell Lab Syst
.
2017
;
171
:
277
–
85
.
51.
Право
CW
,
Чен
Y
,
Ши
3
W 90etal
.
voom: точные веса открывают доступ к инструментам анализа линейных моделей для подсчета прочтений РНК-секвенций
.
Геном Биол
.
2014
;
15
:
R29
.
52.
Смит
ГК
.
Линейные модели и эмпирические байесовские методы для оценки дифференциальной экспрессии в экспериментах с микрочипами
.
Stat Appl Genet Mol Biol
.
2004
;
3
:
Статья3
.
53.
Ценг
ГК
,
Гош
Д
,
Фейнгольд
Всесторонний обзор литературы и статистические соображения для метаанализа микрочипов
.
Рез. нуклеиновых кислот
.
2012
;
40
(
9
):
3785
–
99
.
54.
Лук-порей
JT
.
svaseq: удаление пакетных эффектов и других нежелательных шумов из данных секвенирования
.
Рез. нуклеиновых кислот
.
2014
;
42
(
21
):
e161
.
55.
Мартин-Фернандес
J
,
Барсело-Видаль
C
,
Павловски0003
В
и др. .
Меры различия для композиционных данных и методы иерархической кластеризации
. В:
Труды IAMG
. об.
98
;
1998
:
526
–
531
.
56.
Толосана Дельгадо
R
.
Использование и неправильное использование данных о составе в седиментологии
.
Геол отложений
.
2012
;
280
(
S.I
):
60
–
79
.
57.
Rivera-Pinto
J
,
Egozcue
JJ
,
Pawlowsky-Glahn
V
, et al. .
Весы: новый взгляд на анализ микробиома
.
мСистемы
.
2018
;
3
(
4
):
e00053
–
18
.
58.
Куинн
TP
,
Эрб
I
.
Использование весов для разработки характеристик классификации биомаркеров здоровья: новый подход к выбору весов
.
биоРксив
.
2019
, дои:
10.1101/600122
.
59.
Calle
ML
.
Статистический анализ данных метагеномики
.
Геномикс Информ
.
2019
;
17
(
1
):
e6
.
60.
Gloor
ГБ
,
Маклейм
JM
,
Ву
М
3
и др. .
Неопределенность состава не следует игнорировать при высокопроизводительном анализе данных секвенирования
.
Австрийский J Stat
.
2016
;
45
:
73
–
87
.
61.
Цзян
L
,
Schlesinger
F
,
Davis
3
CA. .Синтетические вводные стандарты для экспериментов с секвенированием РНК
.
Геном Res
.
2011
;
21
(
9
):
1543
–
51
.
62.
Stämmler
F
,
Gläsner
J
,
Hiergeist
A
, et al. .
Корректировка профилей микробиома с учетом различий в микробной нагрузке бактериями
.
Микробиом
.
2016
;
4
(
1
):
28
.
63.
Ткач
А
,
Хортала
М
,
Пул
PS.
Абсолютный количественный анализ микробиоты в пробах окружающей среды
.
Микробиом
.
2018
;
6
:
110
.
64.
Риссо
D
,
Нгаи
J
,
Скорость
TP .Нормализация данных секвенирования РНК с использованием факторного анализа контрольных генов или образцов
.
Нат Биотехнолог
.
2014
;
32
:
896
.
65.
Маргера
S
,
Шмидт
А
,
Кодлин
3
S et al. .Количественный анализ транскриптомов и протеомов делящихся дрожжей в пролиферирующих и покоящихся клетках
.
Сотовый
.
2012
;
151
(
3
):
671
–
83
.
66.
Чен
К
,
Ху
Z
,
Ся
90etal 3 ,Z 90et .
Упускаемый из виду факт: фундаментальная потребность в контроле всплесков практически для всех полногеномных анализов
.
Мол Селл Биол
.
2016
;
36
(
5
):
662
–
67
.
67.
Хардвик
SA
,
Чен
WY
,
Вонг
3 T 9 .Сплайсированные синтетические гены в качестве внутреннего контроля в экспериментах по секвенированию РНК
.
Естественные методы
.
2016
;
13
(
9
):
792
–
8
.
68.
Deveson
IW
,
Chen
WY
,
Wong
T
, и др. .
Представление генетической изменчивости с помощью синтетических стандартов ДНК
.
Естественные методы
.
2016
;
13
(
9
):
784
–
91
.
69.
Симс
DJ
,
Harrington
RD
,
Polley
EC
и др. .
Материалы на основе плазмид в качестве мультиплексных контролей качества и калибраторов для клинических анализов секвенирования следующего поколения
.
Дж Мол Диагн
.
2016
;
18
(
3
):
336
–
49
.
70.
Пол
L
,
Кубала
P
,
Horner
G
, и др. .
SIRV: варианты РНК с шипами в качестве внешних контролей изоформ при секвенировании РНК
.
биоРксив
.
2016
, doi:
10.1101/080747
.
71.
Хардвик
SA
,
Чен
WY
,
Вонг
T
и др. .
Сообщества синтетических микробов обеспечивают внутренние справочные стандарты для секвенирования и анализа метагенома
.
Нац Коммуна
.
2018
;
9
(
1
):
3096
.
72.
AlJanahi
AA
,
Danielsen
M
,
Данбар
30234 .
Введение в анализ данных секвенирования одноклеточной РНК
.
Mol Ther Methods Clin Dev
.
2018
;
10
:
189
–
96
.
73.
Лун
ATL
,
МакКарти
DJ
,
Мариони
0 JC
0
Пошаговый рабочий процесс для низкоуровневого анализа данных секвенирования РНК одиночных клеток с помощью Bioconductor
.
F1000Res
.
2016
;
5
:
2122
.
74.
LUN
ATL
,
Calero-Nieto
FJ
,
Haim-Vilmovsky
L
, et al. .
Оценка надежности нормализации всплесков для анализа данных секвенирования одноклеточной РНК
.
Геном Res
.
2017
, doi:
10.1101/гр.222877.117
.
75.
Kolodziejczyk
A
,
KIM
JK
,
Svensson
V
, et al. .
Технология и биология секвенирования одноклеточной РНК
.
Мол Ячейка
.
2015
;
58
(
4
):
610
–
20
.
76.
Van Den Berge
K
,
Perraudeau
F
,
Soneson
C
, et al. .
Наблюдательные гири открывают инструменты секвенирования РНК для нулевого надувания и одноклеточных приложений
.
Геном Биол
.
2018
;
19
(
1
):
24
.
77.
Martín-Fernández
JA
,
Barceló-Vidal
C
,
Pawlowsky-Glahn
V
4.
Работа с нулями и пропущенными значениями в наборах композиционных данных с использованием непараметрического вменения
.
Мат Геол
.
2003
;
35
(
3
):
253
–
78
.
78.
ван ден Бугаарт
KG
,
Толосана-Дельгадо
R
.
Нули, пропуски и выбросы
. В:
Анализ композиционных данных с помощью R. Используйте R!
.
Берлин, Гейдельберг
:
Springer
;
2013
:
209
–
53
.
79.
Куинн
ТП
.
Полевое руководство по композиционному анализу данных any-omics: дополнительные скрипты
.
Зенодо
.
2018
. http://doi.org/10.5281/zenodo.3270954
Примечания автора
Марк Ф. Ричардсон и Тэмсин М. Кроули Внесли равный вклад.
© Автор(ы), 2019. Опубликовано Oxford University Press.
Это статья в открытом доступе, распространяемая в соответствии с лицензией Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), которая разрешает неограниченное повторное использование, распространение и воспроизведение на любом носителе при условии, что оригинальная работа правильно цитируется.
© Автор(ы), 2019. Опубликовано Oxford University Press.
Состав предков — 23andMe International
Современный анализ географического происхождения от 23andMe
Отчет о составе предков 23andMe — это мощная и хорошо протестированная система для
анализ родословной на основе ДНК, и мы считаем, что это устанавливает стандарт строгости в
индустрия генетического происхождения. Мы написали этот документ, чтобы объяснить, как работает наш анализ, и представить
некоторые результаты проверки качества. Примечание. В этом документе описаны особенности текущей версии.
Ancestry Composition, предлагаемой клиентам на платформе V5. Для клиентов на предыдущих
платформы, нажмите здесь.
В вашем отчете о составе предков показан процент вашей ДНК, полученной от каждого из 47 населения. Мы рассчитываем состав вашего предка, сравнивая ваш геном с геномом более 14 000 человек с известным происхождением. Когда сегмент вашей ДНК близко совпадает с ДНК одного из 47 популяций, мы приписываем эту родословную соответствующему сегменту вашей ДНК. Мы рассчитываем происхождение для отдельных сегментов вашего генома по отдельности, а затем сложить их вместе для вычисления ваш общий родовой состав.
Если у вас есть вопросы, задайте их в сообществе 23andMe или свяжитесь с нами. Покупатель Уход. Вы также можете прочитать нашу технический документ об алгоритме Ancestry Composition.
Основы
Варианты ДНК встречаются с разной частотой в разных местах по всему миру, и каждый маркер
имеет свой характер географического распространения. Алгоритм составления предков 23andMe
объединяет информацию об этих паттернах с уникальным набором аллелей ДНК в вашем геноме, чтобы
оценить свою генетическую родословную.
Вот пример гаплогруппы, особого вида ДНК-маркера, который иллюстрирует эту идею. Этот карта показывает частоту материнской гаплогруппы H по всему миру. Гаплогруппа H очень распространена в Европе, также встречается в Африке и Азии и редко встречается у жителей Австралии или Америка.
Мировое распространение материнской гаплогруппы HСвязь между этим маркером и географическим положением работает двумя способами. Если вы знаете, что у вас есть Европейское происхождение, мы знаем, что у вас есть неплохая вероятность того, что у вас есть гаплогруппа H. И если у вас есть гаплогруппы H, мы знаем, что ваша генетическая история, вероятно, включает по крайней мере одного европейского предка.
Хотя мы не можем определить вашу родословную с большой точностью на основе этого единственного маркера ДНК, мы измеряем
сотни тысяч маркеров ДНК на платформе 23andMe. Если мы объединим свидетельства многих
маркеры, каждый из которых предлагает немного информации о том, откуда вы в мире, мы
может составить четкую общую картину.
Недостаток № 1: люди обычно имеют несколько предков
Если бы вся ваша ДНК происходила из одного места в мире, выяснить, откуда вы родом, было бы легкий. Недавние исследования показали, что для европейца, вся семья которого происходит из одного и того же места, генетический анализ может определить местонахождение их прародины в пределах диапазона около 100 км!
Но предки большинства людей происходят из многих мест. Техническое слово для этого примесь —генетическое смешение ранее обособленных популяций. За например, люди европейского происхождения обычно имеют предки со всей Европы, и У латиноамериканцев обычно есть предки из Америки, Европы и иногда Африки.
Наш алгоритм состава предков решает проблему примеси, разрушая ваши хромосомы.
в короткие соседние окна, как товарные вагоны в поезде. Эти окна настолько малы, что
в целом безопасно предположить, что вы унаследовали всю ДНК в любом данном окне от одного предка
много поколений назад.
Недостаток № 2: мы не знаем, какая ДНК происходит от какого родителя
Вспомните, что для каждой из ваших 23 пар хромосом одна хромосома в каждой паре исходит от вашего мама, а другой от твоего папы. Чипы для генотипирования не захватывают информация о том, какие маркеры произошли от какого родителя.
Вот краткий пример, чтобы проиллюстрировать этот момент. Скажем, на коротком отрезке хромосомы 1 вы унаследовали следующие генотипы по трем последовательным ДНК-маркерам:
от папы: A-T-C
от мамы: G-T-A
Когда мы посмотрим на ваши необработанные данные 23andMe в этом месте на хромосоме 1, мы увидим следующее:
Вы: A/G - T/T - A/C
Генотипы, в которых вы унаследовали разные варианты от мамы и папы — в данном случае
маркеры на концах — перепутаны. Есть два возможных «гаплотипа», которые согласуются друг с другом.
с необработанными данными, и мы не знаем, какая из них является вашей настоящей последовательностью ДНК. Возможно:
А-Т-А
Г-Т-С
что оказывается неправильным, или это может быть:
A-T-C
G-T-A
какой правильный. Технический термин для определения того, какие аллели находятся на одной и той же хромосоме. вместе это фазировка . Данные ДНК, подобные нашим необработанным данным, называются бесфазный .
И что? Это важно, потому что мы можем узнать больше из длинных серий многих маркеров ДНК вместе, чем мы может учиться только по отдельным ДНК-маркерам. В приведенном выше примере комбинация A-T-C, как правило, скажет больше о вашей родословной, чем А, Т и Скажем, когда их рассматривают по отдельности. К счастью, мы можем использовать статистические методы для оценки фазирования ваших хромосом. После поэтапной обработки необработанных данных Алгоритм Ancestry Composition вычисляет родословную отдельно для каждой поэтапной хромосомы.
Подготовка: определение популяций предков
Подготовка 1: наборы данных
Алгоритм состава предков вычисляет вашу родословную, сравнивая ваш геном с геномами
людей, чьи предки мы уже знаем. Чтобы это работало, нам нужно много справочных данных! Наш эталонных наборов данных включают генотипы 14 812 человек, которые были выбраны в целом
чтобы отразить население, которое существовало до того, как трансконтинентальные путешествия и миграция стали обычным явлением (на
не менее 500 лет назад). Однако, поскольку в разных частях света есть свои уникальные
демографической истории, некоторые результаты Состава предков могут отражать родословную из гораздо более широкой
временное окно, чем за последние 500 лет.
Клиенты составляют львиную долю эталонных наборов данных, используемых Ancestry Composition. Когда
Участник исследования 23andMe рассказал нам, что у них четверо бабушек и дедушек, все они родились в одном городе.
страны — и население этой страны не испытывало массовых миграций в последние
несколько сотен лет, как это произошло по всей Америке и, например, в Австралии, — что
человек становится кандидатом на включение в справочные данные. Мы отфильтровываем все, кроме одного из любых
набор близкородственных людей, поскольку включение близкородственных родственников может исказить результаты. И мы удаляем выбросы: люди, чье генетическое происхождение не совпадает с данными их опроса.
ответы. Чтобы обеспечить репрезентативный набор данных, мы применяем агрессивную фильтрацию — почти десять процентов
Кандидаты на эталонные наборы данных не проходят отбор.
Мы также используем общедоступные справочные наборы данных, в том числе Человеческий геном Проект разнообразия, HapMap и 1000 Геномный проект. Наконец, мы включили данные из проектов, спонсируемых 23andMe, которые обычно сотрудничество с академическими исследователями. Мы выполняем одинаковую фильтрацию на общедоступных и справочные данные о совместной работе, которые мы делаем с данными клиентов 23andMe.
Подготовка 2: Выбор популяции
47 популяций по составу предков определяются генетически сходными группами людей с
известное происхождение. Мы выбираем популяции по составу предков, изучая эталонные наборы данных,
выбор популяций-кандидатов, которые кажутся сгруппированными вместе, а затем оценка того, можем ли мы
различать эти группы на практике. Используя этот метод, мы уточнили ссылку-кандидата
населения, пока мы не пришли к набору, который работает хорошо.
Вот пример одного из диагностических графиков, которые мы используем для выбора популяций. Геномы в
Европейские эталонные наборы данных построены с использованием анализа основных компонентов, который показывает их
общее генетическое расстояние друг от друга. Каждая точка на графике представляет одного человека, и мы
пометили точки разными символами и цветами в зависимости от их известного происхождения. Ты можешь видеть
что люди из одной популяции (помеченные одним и тем же символом) имеют тенденцию группироваться вместе. Немного
популяции, такие как финны (синие треугольники слева), относительно изолированы от
другие популяции. Поскольку финны настолько генетически различны, у них есть своя собственная референция.
населения в составе предков. Однако большинство групп населения на уровне страны пересекаются с некоторыми
степень. В этих случаях мы экспериментировали с различными группами населения на уровне страны, чтобы
найти комбинации, которые мы могли бы различить с высокой уверенностью.
Некоторых генетических предков трудно различить по своей природе, потому что люди в этих регионах смешанные на протяжении всей истории или имеющие общую историю. По мере того, как мы будем получать больше данных, популяции станут легче различить, и мы сможем сообщить о большем количестве популяций в составе предков. отчет.
Противостояние предвзятости
Исторически биомедицинские исследования были непропорционально сосредоточены на участниках европейского происхождения.
В свете этого неравенства исследовательская группа 23andMe постоянно работает над получением новых данных от
разнообразное население. Наша миссия в 23andMe — помочь людям получить доступ, понять и извлечь выгоду из
геном человека. Лучший способ сделать это для малообеспеченных слоев населения — включить их
генетические данные в наших исследованиях и в наших функциях предков - максимизируя
детализация состава предков для всех наших клиентов и помощь в борьбе с несоответствиями в
генетическая наука. Мы активно работали над тем, чтобы уменьшить предвзятость в генетических исследованиях, инициировав
таких проектов, как Глобальный генетический проект,
Африканский генетический проект,
Программа сотрудничества в области народонаселения и наша
Финансируемый NIH генетический ресурс здоровья для афроамериканцев.
Генетическая информация, которую мы собираем в рамках этих и подобных им инициатив, поможет
улучшить такие функции, как состав предков, и принести пользу научному сообществу в целом.
Алгоритм композиции предков
Обзор
Алгоритм Ancestry Composition состоит из четырех отдельных шагов.
Во-первых, мы используем вычислительный метод для оценки фазирования ваших хромосом, то есть для
определить вклад в ваш геном каждого из ваших родителей. Далее разбиваем
хромосом на короткие окна, и мы сравниваем вашу последовательность ДНК в каждом окне с
соответствующую ДНК в наших справочных наборах данных. Мы маркируем вашу ДНК родословной, ссылка на которую
ДНК очень похожа, а затем мы вычисляем эти назначения, чтобы «сгладить» их. Каждый шаг в этом процессе описан более подробно в следующих разделах.
Шаг 1: Поэтапность
Вспомните морщину № 2 выше. Для каждого клиента мы измеряем набор генотипов (пар аллелей). Но на самом деле нам нужна пара гаплотипов для каждой хромосомы. То есть мы хотим вычислить из серии аллелей, присутствующих на каждой из ваших двух копий, например, хромосомы 7: один вы получили от своей матери и один вы получили от своего отца. Для этого мы сначала построить очень большую «справочную панель фазирования», используя данные сотен тысяч клиенты. Затем мы используем Eagle (Loh et al., 2016), чтобы объединить этих людей. Орел использует сложную статистику и очень умный алгоритм для этого. Как только мы поэтапно это большое количество клиентов, мы можем использовать полученную информацию для эффективного фаза новых клиентов.
Шаг 2: Классификация окон
После фазирования ваших хромосом мы сегментируем их на последовательные окна, содержащие ~300 генетических
маркеры каждый. Мы измеряем от 7400 до 45000 маркеров на хромосому, что соответствует от 24 до
149 окон, в зависимости от длины хромосомы. Мы рассматриваем каждое окно по очереди и сравниваем ваши
ДНК в эталонные наборы данных, чтобы определить, какая родословная наиболее точно соответствует вашей ДНК.
Есть много способов определить происхождение сегментов ДНК на основе справочных данных, и мы попробовали несколько. Наиболее эффективным вариантом был известный инструмент классификации, называемый опорным вектором. машина или SVM. SVM может «изучить» различные классификации предков на основе набора обучающих данных. примеры, а затем назначать новые сегменты ДНК изученной категории.
В случае Ancestry Composition мы обучаем SVM с эталонными последовательностями ДНК и сообщаем ему, какие
популяция предков, из которой эти последовательности. Затем, когда мы посмотрим на ДНК клиента 23andMe
с неизвестным происхождением (как вы), мы можем попросить SVM классифицировать вашу ДНК для нас на основе
справочные наборы данных.
Мы выбрали алгоритм Ancestry Composition, основанный на SVM, потому что он работает лучше всех. методики, которые мы пробовали. Кроме того, SVM очень быстры, что очень важно для крупного и растущего база данных.
Шаг 3: Сглаживание
SVM классифицирует каждое окно вашего генома независимо, создавая «первую черновую» версию генома. результат вашего происхождения. Мы используем другой вычислительный процесс, называемый сглаживающим , чтобы сгладить этот необработанный вывод SVM. Сглаживатель использует версию известного математического инструмента под названием Скрытая марковская модель для исправления или «сглаживания» двух видов ошибок. Используются скрытые марковские модели. для анализа последовательных данных, таких как биологические последовательности или записанная речь. Как Например, предположим, что у нас есть три популяции предков: X, Y и Z. Пример выходных данных из SVM может выглядеть так:
Хромосома 1, родитель 1: X - X - X - Z - Z - Z - Y - Z
Хромосома 1, родитель 2: Z - Z - Z - X - X - X - X - X
Первый вид ошибок, которые корректирует сглаживатель, — это необычное задание в середине цикла. подобные задания. В первой строке выше есть серия Z, прерванная одной Y:
Z-Z-Z-Y-Z. Возможно, что одинокий Y был близким вызовом
между Y и Z, что пошло не так. Если бы это было так, сглаживатель мог бы исправить это на
З-З-З-З-З.
Второй тип ошибок, которые исправляет сглаживатель, возникает из-за шага фазирования. Алгоритмы фазирования могут совершать ошибки, известные как ошибки переключения , когда они смешивают ДНК одного родителя с тем другого. Smoother может переключать назначения родословной между вашей матерью и вашим отцу, если он обнаружит одну из этих ошибок. В этом примере может возникнуть ошибка переключения после четвертое окно. Если бы переключатель был перевернут, то серии X и серии Z остались бы вместе. В нашем упрощенном примере сглаживатель может вывести что-то вроде этого:
Хромосома 1, родитель 1: Z - Z - Z - Z - Z - Z - Z - Z
Хромосома 1, родитель 2: X - X - X - X - X - X - X - X
Этот пример иллюстрирует назначение сглаживателя. Но с реальными данными картина намного
грязнее, и ответы редко бывают такими чистыми. Таким образом, вместо того, чтобы назначать одно происхождение каждому
окно, как мы сделали в этом примере, более гладкие оценки вероятностей каждого
Родословная Состав населения, соответствующий каждому окну ДНК. На следующем рисунке показан бетон
пример:
Это результат более гладкого анализа одной копии хромосомы 2. Начиная слева,
есть короткая полоса розового, затем более широкая полоса зеленого, затем еще одна полоса розового. На этом графике
розовый - это цвет африканцев к югу от Сахары, а зеленый - коренной американец.
ось Y проходит от 0 до 100 процентов, и она показывает вероятность того, что ДНК в этой области
хромосома происходит от каждой популяции состава предков. Эти розовые и зеленые области заполняют
всему вертикальному пространству графика, а это значит, что мы на 100 процентов уверены, что ДНК в
эти регионы имеют генетическое происхождение африканцев к югу от Сахары и коренных американцев соответственно.
Следующая область справа — между позициями 50 и 100 по оси x — представляет собой участок
разноцветный синий. Самая толстая полоса внизу темно-бирюзовая, это цвет британцев и
Ирландский. Этот сегмент ДНК имеет где-то между 50-процентной и 60-процентной вероятностью
отражая британское и ирландское происхождение. Другие оттенки синего показывают, что один и тот же сегмент ДНК
также может отражать итальянское, иберийское или французское и немецкое происхождение. Если вы думаете о
приведенный выше пример гаплогруппы, этот результат имеет смысл: совпадение ДНК-маркера является нормальным
эталонная ДНК из множества мест, даже если в одних местах она совпадает лучше, чем в других. В этом
Например, результат показывает, что этот сегмент ДНК соответствует эталонной ДНК со всей Европы. Мы можем
можно с уверенностью заключить, что этот отрезок ДНК отражает европейское происхождение, но доказательства
недостаточно сильны, чтобы с высокой уверенностью отнести его к одному конкретному региону Европы.
Шаг 4: Агрегация и отчетность
Последний шаг — обобщить результаты и отобразить их в вашей Хромосомной картине. Как мы сделать это, чтобы применить порог к графику вероятности, как на этом рисунке:
Применение порога к вероятностям назначения состава предковГоризонтальная линия на этом изображении указывает 70-процентный доверительный порог, который мы будем использовать. для этого примера. Вы можете просмотреть свою собственную Хромосомную картину с различными порогами достоверности, от 50 процентов (спекулятивных) до 90 процентов (консервативный).
Мы просматриваем всю хромосому и спрашиваем, есть ли у какого-либо предка предполагаемая вероятность
превышение указанного порога (в данном случае 70 процентов). В этом примере, за исключением
синей европейской полосы, оценки предков превышают 70 процентов по большинству
хромосома. Каждый регион вносит свой вклад в ваш общий состав предков пропорционально его
размер: например, зеленый сегмент коренных американцев ближе к концу этого графика составляет около 0,26
процентов всего генома. Несмотря на то, что существует некоторая вероятность того, что сегмент происходит из
различное население, доля коренных американцев превышает 70-процентный порог, и поэтому мы
добавьте 0,26 процента коренных американцев к общему составу предков при этом пороге.
В случае европейского сегмента ни одно происхождение не превышает 70-процентного порога, поэтому мы не приписывайте эту ДНК каким-либо мелкозернистым предкам. Вместо этого мы ссылаемся на нашу иерархию предки. Существует «широкая североевропейская» родословная, которая включает в себя четыре высококлассных предки: британцы и ирландцы, скандинавы, финны, французы и немцы. Если при сложении вклад каждой из этих подгрупп, общий вклад в широкое североевропейское превышает 70-процентный порог, тогда мы будем сообщать о регионе как о широко распространенном северноевропейском.
В этом примере эталонная популяция в целом для Северной Европы по-прежнему не превышает 70 человек.
процентный порог, но объединенные вероятности всего европейского населения. Так это
региону присвоено «широкоевропейское» происхождение.
Мы используем широкие категории состава предков, чтобы не делать предположений о вашем происхождении, когда ваша ДНК соответствует нескольким различным популяциям на уровне страны. В регионах, где нет родословная, в том числе широкие предки, превышает указанный порог, мы сообщаем «Неназначенное» происхождение. Вы можете увидеть всю иерархию предков в вашей композиции предков. отчет, нажав «Просмотреть все протестированные группы населения».
Связь с близкой семьей
Состав предков еще более эффективен, если у вас есть биологический родитель, который также находится в База данных 23andMe. Нажмите здесь, чтобы узнать больше о связи с семьей и друзья.
Ваша связь с биологическим родителем значительно упрощает вычислительную задачу
выяснение того, какую ДНК вы получили от какого родителя (см. Шаг 1: Фазирование). Это может
перевести в лучшие результаты композиции предков, в том смысле,
что вы могли бы увидеть больше отнесений к предкам с высоким разрешением: больше скандинавских, меньше
Североевропейский.
Почему это? Помните, что чем мягче, тем больше будет ваша окончательная композиция предков. оценка — должна исправлять два вида ошибок: ошибки вдоль хромосомы и ошибки между хромосомы. Когда ваши хромосомы фазируются с использованием генетической информации от ваших родителей, ошибки между хромосомами (ошибки переключения) встречаются крайне редко, поэтому чем ровнее, тем увереннее.
Если вы соединитесь с одним или обоими вашими биологическими родителями, вы получите дополнительный результат. Ты будешь
возможность увидеть представление «Родительское наследство», которое показывает вклад вашей матери в вашу родословную
с одной стороны, и вклад вашего отца в вашу родословную с другой. Мы не можем предоставить это
просмотреть, если у вас нет подключенного родителя, потому что нам нужен хотя бы один из ваших родителей, чтобы ориентироваться
результаты, достижения. Вот пример того, что вы можете узнать из просмотра наследования: скажите, что ваша родословная
Композиция включает в себя небольшое количество еврейского происхождения ашкенази. Когда вы смотрите на свое наследство
View, вы сможете увидеть, от какого родителя вы его унаследовали.
Тестирование и проверка
Ancestry Composition включает в себя множество шагов, и каждый шаг необходимо протестировать. Мы обсудили несколько этих тестов уже при объяснении нашего алгоритма. В этом разделе мы хотим поделиться некоторыми результаты теста, чтобы понять, насколько хорошо работает Ancestry Composition. Этот раздел посвящен последний тест, который мы проводим, потому что он объединяет производительность каждого из шагов в общую картина.
В этом тесте рассматриваются два классических показателя производительности модели, точность и отзыв . Это стандартные измерения, которые исследователи используют для проверки того, насколько хорошо
работает система прогнозирования. Точность отвечает на вопрос: «Когда система предсказывает, что часть
ДНК исходит от популяции А, как часто ДНК на самом деле принадлежит популяции А?» Вспомните ответы на вопросы. вопрос «Сколько фрагментов ДНК, которые на самом деле принадлежат популяции А, как часто система
правильно предсказать, что они из популяции А?"
Существует компромисс между точностью и отзывом, поэтому мы должны найти баланс между ними. А высокоточная малоотзывная система будет крайне требовательна к присвоению, скажем, скандинавских родословная. Система назначит ДНК как скандинавскую только тогда, когда будет очень уверена. Что будет дают высокую точность, поскольку присвоение скандинавского языка почти всегда правильное, но низкий отзыв, потому что многие истинные скандинавские предки остаются неназначенными.
С низкоточной системой с высоким откликом существует противоположная проблема. В этом случае система
свободно указывает скандинавское происхождение. Каждый раз, когда фрагмент ДНК может быть скандинавским, он
присваивается это происхождение. Это обеспечит высокий уровень отзыва, так как большая часть подлинной скандинавской ДНК будет
помечены соответствующим образом, но с низкой точностью, потому что нескандинавская ДНК часто будет неправильно
помечены как скандинавские.
Идеальная система обладает как высокой точностью, так и высоким откликом, но в реальной жизни это может быть невозможно. Давайте посмотрим, как Ancestry Composition работает с этими показателями. Для этого теста контроля качества мы установили помимо 20 процентов справочной базы данных, примерно 3000 человек известного происхождения. Мы обучили и запустили весь конвейер Ancestry Composition на остальных 80 процентах эталона лица. Затем мы относились к 20% «удерживающихся» как к новым клиентам 23andMe. и использовали наш конвейер Ancestry Composition для расчета их предков. Поскольку мы знаем эти истинные предки людей, мы можем проверить, насколько точны результаты их состава предков. Мы провели этот тест пять раз каждый с различными минимальными доверительными порогами, с разными 20 процентов выдерживали каждый раз, а затем усредняли по пяти тестам, чтобы получить следующие результаты: (показано здесь для минимального доверительного порога 50%, который является значением по умолчанию для результатов, показанных клиенты):
| Население | Точность (%) | Отзыв (%) | |||
|---|---|---|---|---|---|
| Sub-Saharan African | 99 | 98 | |||
| West African | 98 | 99 | |||
| Senegambian & Guinean | 94 | 95 | |||
| Ghanaian, Liberian & Sierra Leonean | 96 | 87 | |||
| Нигериец | 91 | 99 | |||
| Northern East African | 97 | 92 | |||
| Sudanese | 94 | 82 | |||
| Ethiopian & Eritrean | 93 | 97 | |||
| Somali | 99 | 91 | |||
| Конголезцы и южно-восточноафриканские | 97 | 99 | |||
| Ангольцы и конголезцы | 9 908 8 989|||||
| Southern East African | 95 | 96 | |||
| African Hunter-Gatherer | 98 | 86 | |||
| Indigenous American | 98 | 93 | |||
| East Asian | 98 | 99 | |||
| North Asian | 57 | 90 | |||
| Siberian | 90 | 93 | |||
| Manchurian & Mongolian | 38 | 80 | |||
| Chinese | 97 | 98 | |||
| Northern Chinese & Tibetan | 82 | 95 | |||
| Southern Chinese & Taiwanese | 88 | 73 | |||
| South Китайский | 88 | 92 | |||
| Вьетнамский | 96 | 96 | |||
| Филиппинский и австронезийский 9 | |||||
| 88 | |||||
| Indonesian, Khmer, Thai & Myanma | 94 | 60 | |||
| Chinese Dai | 89 | 97 | |||
| Japanese | 100 | 99 | |||
| Korean | 96 | 99 | |||
| Европейский | 99 | 99 | |||
| Северный европейский0273 | 95 | 88 | |||
| Finnish | 94 | 95 | |||
| French & German | 79 | 80 | |||
| Scandinavian | 91 | 90 | |||
| Southern European | 90 | 90 | |||
| Греческий и балканский | 89 | 80 | |||
| Испанский и португальский | 90 | 8 80271 | Italian | 87 | 86 |
| Sardinian | 85 | 93 | |||
| Eastern European | 86 | 88 | |||
| Ashkenazi Jewish | 99 | 98 | |||
| Western Asian и Северная Африка | 95 | 95 | |||
| Северо-Западная Азия | 83 | 91 | |||
| Кипр | |||||
| 93 | |||||
| Anatolian | 89 | 64 | |||
| Iranian, Caucasian & Mesopotamian | 66 | 93 | |||
| Arab, Egyptian & Levantine | 94 | 85 | |||
| Peninsular Arab | 88 | 69 | |||
| Levantine | 93 | 69 | |||
| Egyptian | 78 | 88 | |||
| Coptic Egyptian | 91 | 95 | |||
| North African | 97 | 89 | |||
| Central & South Asian | 98 | 96 | |||
| Central Asian | 87 | 44 | |||
| Северо-Индийский и Пакистанский | 81 | 87 | |||
| Бенгальский и Северо-Восточный Индийский | 92 | 94 | |||
| Гуджа0273 | 98 | 97 | |||
| Southern Indian Subgroup | 93 | 84 | |||
| Southern Indian & Sri Lankan | 78 | 93 | |||
| Malayali Subgroup | 93 | 75 | |||
| Меланезийский | 98 | 96 |
Эта таблица показывает, что наши показатели точности высоки по всем направлениям, в основном выше 90 процентов, и
редко опускаясь ниже 75 процентов. Это означает, что когда система присваивает происхождение части
ДНК, это определение, скорее всего, будет точным. Вы также можете видеть, что по мере продвижения вверх от
субрегиональный уровень (например, британский и ирландский) на региональный уровень (например, североевропейский) на
континентальном уровне (например, европейском) точность приближается к 100 процентам.
Важно понимать, что плохой отзыв не означает плохих результатов. Немного такие популяции, как сардинцы, просто трудно отличить от других. Когда состав предков не может присвоить сардинскую ДНК, это не означает, что ДНК неправильно приписана чему-то иначе, как итальянский. Если бы это было так, то итальянское население имело бы плохую точность. Вместо, Состав предков часто относит сардинскую ДНК к широко южноевропейскому или широко распространенному Европейские популяции.
Композиция «Будущее предков»
Ancestry Composition имеет модульную конструкцию. Это было сделано намеренно, потому что это позволяет нам улучшить
отдельные компоненты системы, такие как эталонная база данных фазирования Eagle или SVM. эталонные группы населения, не влияя на какие-либо другие этапы конвейера анализа.
Мы надеемся регулярно обновлять Ancestry Composition. Когда мы улучшаем какой-то компонент системы или обновите эталонные наборы данных, ваши результаты будут автоматически обновлены. Вы будете в состоянии чтобы увидеть список этих обновлений в журнале изменений в нижней части вашей композиции предков. Научные детали.
Обновлено в августе 2022 г.
Как выбрать правильную диаграмму для ваших данных
Как выбрать правильную диаграмму или график для ваших данных?
Если у вас есть данные, которые вы хотите визуализировать, убедитесь, что вы используете правильные диаграммы. Хотя ваши данные могут работать с несколькими типами диаграмм, вы должны выбрать тот, который обеспечит четкость и точность вашего сообщения. Помните, что данные ценны только в том случае, если вы знаете, как их визуализировать и дать контекст.
Мы дадим вам обзор различных типов диаграмм и объясним, как выбрать правильный.
О чем рассказывают ваши данные?
Перед созданием диаграммы важно понять, зачем она вам нужна. Диаграммы, карты и инфографика помогают людям понимать сложные данные, находить закономерности, выявлять тенденции и рассказывать истории. Подумайте о сообщении, которым вы хотите поделиться со своей аудиторией.
Следуйте рекомендациям по составлению графиков. Ваши числа должны складываться, и диаграммы должны быть соответственно масштабированы. Что бы вы хотели показать? Существует четыре основных типа диаграмм:
Источник: The Extreme Presentation Method
Узнайте, как использовать передовой опыт сторителлинга для создания потрясающих изображений и эффектных презентаций, привлекающих аудиторию.
СравнениеСравнительные диаграммы используются для сравнения одного или нескольких наборов данных. Они могут сравнивать предметы или показывать различия во времени.
Взаимосвязь Диаграммы взаимосвязей используются для отображения связи или корреляции между двумя или более переменными.
Диаграммы композиции используются для отображения частей целого и изменения во времени.
РаспределениеДиаграммы распределения используются, чтобы показать, как переменные распределяются во времени, помогая выявить выбросы и тенденции.
Выбор правильного типа диаграммы
Спросите себя, сколько переменных вы хотите отобразить, сколько точек данных вы хотите отобразить и как вы хотите масштабировать свою ось.
Линейные, гистограммы и гистограммы отображают изменения с течением времени. Пирамиды и круговые диаграммы отображают части целого. В то время как точечные диаграммы и древовидные карты полезны, если у вас есть много данных для визуализации.
Типы диаграмм
Линейные диаграммы
Линейная диаграмма показывает тенденции или изменения во времени. Линейные диаграммы можно использовать для отображения взаимосвязей в непрерывном наборе данных, и их можно применять к широкому спектру категорий, включая ежедневное количество посетителей сайта или колебания цен на акции.
Рекомендации по созданию линейных диаграмм:
Четко обозначьте свои оси. Убедитесь, что зритель знает, что он оценивает.
Удалите отвлекающие элементы диаграммы. Сетки, различные цвета и громоздкие легенды могут отвлекать зрителя от быстрого просмотра общей тенденции.
Увеличьте масштаб оси Y, если ваш набор данных начинается выше нуля. В некоторых случаях изменение масштаба оси Y упрощает задачу.
- Избегайте сравнения более 5-7 строк. Вы не хотите, чтобы ваша диаграмма была загромождена или ее было трудно читать. Визуализируйте данные, необходимые для рассказа вашей истории, не более того.
Круговые диаграммы
Круговая диаграмма является одним из наиболее часто используемых и ненавистных типов диаграмм всех времен. Круговые диаграммы используются для отображения частей целого. Круговая диаграмма представляет числа в процентах, а общая сумма всех разделенных сегментов равна 100 процентам.
Рекомендации по созданию круговых диаграмм:
Убедитесь, что ваши сегменты в сумме составляют 100. Звучит очевидно, но это распространенная ошибка.
Содержите его в чистоте и последовательности. Сравните всего несколько категорий, чтобы донести свою точку зрения. Если секторы круговой диаграммы имеют примерно одинаковый размер, рассмотрите возможность использования гистограммы или гистограммы.
- Не используйте трехмерные изображения и не наклоняйте круговую диаграмму. Это часто делает ваши данные нечитаемыми, потому что зритель пытается быстро сравнить ракурсы.
Гистограммы и гистограммы
Гистограммы и гистограммы используются для сравнения различных элементов. Столбцы на столбчатой диаграмме расположены вертикально, а столбцы на столбчатой диаграмме - горизонтально. Гистограммы обычно используются, чтобы избежать беспорядка, когда одна метка данных длинная или если у вас есть более 10 элементов для сравнения. Их легко понять и создать.
Рекомендации по созданию столбчатых и столбчатых диаграмм:
- Начать ось Y с нуля. Наши глаза чувствительны к области столбцов на диаграмме. Если эти полосы усечены, зритель может сделать неверные выводы.
- Пометьте оси. Пометка осей дает контекст для вашего средства просмотра.
- Поместите метки значений на стержни — это помогает сохранить четкие линии длин стержней.
- Избегайте использования слишком большого количества цветов с эффектом радуги. Гораздо лучше использовать один цвет или различные оттенки одного и того же цвета. Вы можете выделить одну полосу, в частности, если это сообщение, которое вы хотите донести.
Карта дерева
Карты дерева показывают части целого. Они отображают иерархическую информацию в виде кластера прямоугольников разного размера и цвета в зависимости от значения данных. Размер каждого прямоугольника представляет количество, а цвет может представлять числовое значение или категорию.
Древовидные карты позволяют просматривать тенденции и быстро проводить сравнения, особенно если один цвет особенно выделяется. В то время как электронные таблицы могут отображать несколько строк данных, древовидные карты могут содержать сотни тысяч элементов в одном организованном отображении, что позволяет легко выявлять закономерности за считанные секунды. Кроме того, если они сделаны правильно, они очень эффективно используют пространство.
Рекомендации по созданию древовидной карты
- Начните с чистых данных и ясного сообщения. Древовидные карты часто могут содержать много данных, поэтому важно точно знать, что вы хотите выделить.
- Используйте яркие, контрастные цвета, чтобы каждую область было легко определить. Но не забывайте избегать «эффекта радуги». Выбирайте цвета с умом.
- Надлежащим образом пометьте каждый регион текстом или цифрами, чтобы зрителю было проще безошибочно оценить вашу древовидную карту.
- Не загромождайте карту дерева слишком большим количеством блоков. Карты дерева могут содержать любое количество блоков, но пространство ограничено! Вы не хотите, чтобы ваша древовидная карта была трудной для чтения.
Диаграмма с двумя осями
С диаграммой с двумя осями вы, по сути, объединяете несколько диаграмм и добавляете вторую ось Y для сравнения. Некоторые члены сообщества визуализации данных скептически относятся к использованию диаграмм с двумя осями, потому что они часто могут сбивать с толку, иметь плохой дизайн и вводить зрителя в заблуждение.
Давайте рассмотрим различные типы диаграмм с двумя осями и лучшие способы их использования:
Столбчатая и линейная диаграмма — Эта диаграмма с двумя осями сочетает в себе столбчатую диаграмму и линейную диаграмму.
Двухлинейный график — Этот двухосевой график сравнивает два линейных графика. При необходимости может быть больше двух строк.
Диаграмма с двумя столбцами — Эта диаграмма с двумя осями показывает два набора данных, отображаемых рядом.
Многоосевая схема — отображает наиболее сложную версию диаграммы с двумя осями. Здесь вы видите три набора данных — с тремя осями Y.
Диаграмма с областями
Диаграммы с областями очень похожи на линейные диаграммы, но с небольшими отличиями. Они могут отображать изменения с течением времени, общие тенденции и непрерывность в наборе данных. Но хотя диаграммы с областями могут функционировать так же, как линейные диаграммы, пространство между линией и осью заполняется, указывая на объем.
Рекомендации по созданию диаграмм с областями
Сделайте его легко читаемым - избегайте окклюзии. Это происходит, когда один или несколько слоев закрывают важную информацию на диаграмме.
Используйте диаграмму с областями с накоплением — если у вас есть несколько наборов данных и вы хотите подчеркнуть отношения части к целому.
Используйте диаграммы с областями, чтобы увидеть более широкую картину. Возьмем, к примеру, население: линейные диаграммы хороши для отображения чистого изменения численности населения с течением времени, а диаграммы с областями хороши для отображения общей численности населения с течением времени.
Пирамидальная диаграмма
Пирамидальная диаграмма (треугольная диаграмма или треугольная диаграмма) — это увлекательный способ визуализации фундаментальных отношений. Они отображаются в виде треугольника, разделенного на горизонтальные секции с категориями, помеченными в соответствии с их иерархией. Они могут быть ориентированы вверх или вниз в зависимости от отношений, которые они представляют. Сложенные слои также могут отображать порядок шагов в конкретном процессе.
Рекомендации по созданию пирамидальных диаграмм
Выберите тему и четко обозначьте подкатегории. Решите, какую информацию вы хотите передать с помощью пирамиды, и четко обозначьте слои.
Организуйте свои подкатегории. Определите порядок и значение каждого раздела в вашей пирамиде.
Организуйте подкатегории на основе их иерархии.
Будьте последовательны — соблюдайте равномерное расстояние между разделами и выбирайте приятную цветовую палитру.
Сведите подкатегории к минимуму. Добавление большого количества слоев и цветов может затруднить чтение вашей пирамиды.
Облако слов
Облака слов (также известные как облака тегов) представляют собой тип взвешенного списка. Облака слов отображают текст шрифтом разного размера, веса или цвета, чтобы показать частоты или категории. Они могут быть расположены в алфавитном порядке или в случайном порядке. Они помогают людям выявлять тенденции и закономерности, которые иначе было бы трудно увидеть.
Рекомендации по созданию облака слов
Предоставление контекста. Облака слов визуально привлекают внимание и предоставляют информацию о частоте, но часто не дают зрителю никакого контекста.
Используйте облака слов, чтобы показать частоту. Не используйте их для отображения сложных тем, таких как бюджет или кризис здравоохранения.
Следите за длиной слова. Более длинные слова занимают больше места и могут ввести в заблуждение.
- Облака
Word отлично подходят для фильтрации и анализа данных.
Не делайте слова слишком похожими по размеру или цвету.
Таблицы
В таблицах данные отображаются в строках и столбцах. Таблицы позволяют легко сравнивать пары связанных значений или отображать качественную информацию (например, квартальные продажи за несколько лет).
Есть несколько причин, по которым вы можете выбрать таблицу, а не график, как правильный способ визуализации данных.
Рекомендации по созданию таблиц отчетов
- Задайте себе вопрос, как будет использоваться ваша таблица, и определите свою аудиторию.
- Рассмотрите возможность удаления линий сетки, чтобы улучшить читаемость.
- Всегда указывайте источник(и) ваших данных
Числа должны быть выровнены по правому краю, потому что это облегчает сравнение. Текст можно выровнять по левому краю, но вы можете отцентрировать его для удобочитаемости.
Используйте цвет или форматирование, чтобы привлечь внимание зрителя к определенным значениям (ячейкам) в таблице.
Определение, модель, анализ и пример
Что такое цепочка создания стоимости?
Цепочка создания стоимости — это бизнес-модель, описывающая весь спектр действий, необходимых для создания продукта или услуги. Для компаний, производящих товары, цепочка создания стоимости включает в себя шаги, которые включают в себя создание продукта от концепции до распределения, а также все промежуточные этапы, такие как закупка сырья, производственные функции и маркетинговая деятельность.
Компания проводит анализ цепочки создания стоимости, оценивая подробные процедуры, задействованные на каждом этапе ее бизнеса. Целью анализа цепочки создания стоимости является повышение эффективности производства, чтобы компания могла создавать максимальную ценность с наименьшими возможными затратами.
Цепочка создания стоимости
Ключевые выводы
- Цепочка создания стоимости — это пошаговая бизнес-модель для преобразования продукта или услуги из идеи в реальность.
- Цепочки создания стоимости помогают повысить эффективность бизнеса, чтобы он мог приносить максимальную пользу при наименьших возможных затратах.
- Конечной целью цепочки создания стоимости является создание конкурентного преимущества для компании за счет повышения производительности при сохранении разумных затрат.
- Теория цепочки создания стоимости анализирует пять основных видов деятельности фирмы и четыре вспомогательных вида деятельности.
Понимание цепочки создания стоимости
Из-за постоянно растущей конкуренции за непревзойденные цены, исключительные продукты и лояльность клиентов компании должны постоянно проверять ценность, которую они создают, чтобы сохранить свое конкурентное преимущество. Цепочка создания стоимости может помочь компании выявить области своего бизнеса, которые являются неэффективными, а затем внедрить стратегии, которые оптимизируют ее процедуры для достижения максимальной эффективности и прибыльности.
В дополнение к тому, чтобы производственная механика была безупречной и эффективной, очень важно, чтобы компании поддерживали клиентов в уверенности и достаточной безопасности, чтобы оставаться лояльными. В этом также может помочь анализ цепочки создания стоимости.
Главной целью цепочки создания стоимости является предоставление максимальной ценности при наименьших затратах для создания конкурентного преимущества.
Фон
Майкл Э. Портер из Гарвардской школы бизнеса представил концепцию цепочки создания стоимости в своей книге 9.0041 Конкурентное преимущество: создание и поддержание превосходной производительности . Он писал: «Конкурентное преимущество нельзя понять, глядя на фирму в целом. Оно проистекает из множества отдельных действий, которые фирма выполняет при разработке, производстве, маркетинге, доставке и поддержке своего продукта».
Другими словами, важно максимизировать ценность на каждом конкретном этапе процессов фирмы.
Компоненты цепочки создания стоимости
В своей концепции цепочки создания стоимости Портер разделяет бизнес-деятельность на две категории: «основную» и «вспомогательную», примеры деятельности которых мы приводим ниже. Конкретные виды деятельности в каждой категории будут варьироваться в зависимости от отрасли.
Основная деятельность
Основные виды деятельности состоят из пяти компонентов, и все они необходимы для создания добавленной стоимости и создания конкурентных преимуществ:
- Входящая логистика включает такие функции, как получение, складирование и управление запасами.
- Операции включают процедуры преобразования сырья в готовую продукцию.
- Внешняя логистика включает деятельность по доставке конечного продукта потребителю.
- Маркетинг и продажи включают стратегии повышения узнаваемости и ориентации на соответствующих клиентов, такие как реклама, продвижение и ценообразование.
- Сервис включает в себя программы обслуживания продуктов и повышения качества обслуживания клиентов, такие как обслуживание клиентов, техническое обслуживание, ремонт, возврат средств и обмен.
Вспомогательная деятельность
Роль вспомогательных мероприятий заключается в том, чтобы помочь сделать основные виды деятельности более эффективными. Когда вы повышаете эффективность любого из четырех вспомогательных действий, это приносит пользу как минимум одному из пяти основных действий. Эти вспомогательные мероприятия обычно обозначаются как накладные расходы в отчете о прибылях и убытках компании:
- Закупки касается того, как компания получает сырье.
- Технологические разработки используется фирмой на стадии исследований и разработок (НИОКР), например при проектировании и разработке производственных технологий и автоматизации процессов.
- Управление человеческими ресурсами (HR) включает в себя наем и удержание сотрудников, которые будут выполнять бизнес-стратегию фирмы и помогать разрабатывать, продвигать и продавать продукт.
- Инфраструктура включает в себя системы компании и состав ее управленческой команды, такие как планирование, бухгалтерский учет, финансы и контроль качества.
Примеры цепочек создания стоимости
Корпорация Starbucks
Starbucks (SBUX) представляет собой один из самых популярных примеров компании, которая понимает и успешно реализует концепцию цепочки создания стоимости. Существует множество статей о том, как Starbucks включает цепочку создания стоимости в свою бизнес-модель.
Торговец Джо
Другим примером является частный продуктовый магазин Trader Joe's, который также получил много отзывов в прессе о своей огромной ценности и конкурентоспособности. Поскольку компания является частной, многие аспекты ее стратегии нам неизвестны. Однако, когда вы входите в магазин Trader Joe's, вы можете легко увидеть примеры бизнеса Trader Joe, которые отражают пять основных видов деятельности в цепочке создания стоимости.
1. Входящая логистика. В отличие от традиционных супермаркетов, Trader Joe's занимается получением товаров, их размещением на полках и инвентаризацией в обычные часы работы магазина. Хотя эта система может свести с ума покупателей, она обеспечивает значительную экономию средств только с точки зрения заработной платы сотрудников. Более того, логистика выполнения этой работы в то время, когда клиенты все еще совершают покупки, посылает стратегическое сообщение о том, что «мы все вместе».
2. Операции. Вот пример того, как компания может творчески применить цепочку создания стоимости. В приведенном выше основном виде деятельности номер два «преобразование сырья в готовый продукт» упоминается как «операционная деятельность». Однако, поскольку преобразование сырья не является аспектом индустрии супермаркетов, мы можем использовать термин «операции» для обозначения любой другой обычной функции продуктового магазина. Итак, давайте заменим «разработкой продукта», так как эта операция имеет решающее значение для Trader Joe's.
Компания тщательно отбирает свои продукты, предлагая товары, которые вы обычно не можете найти где-либо еще. Продукты под собственной торговой маркой составляют более 80% ее предложений, которые также часто имеют самую высокую норму прибыли, поскольку Trader Joe's может эффективно поставлять их в больших объемах. Еще одним важным элементом разработки продуктов для Trader Joe's являются его программы дегустации и партнерские программы с шеф-поваром, которые обеспечивают высокое качество и постоянное совершенствование продукта.
3. Исходящая логистика. Многие супермаркеты предлагают доставку на дом, а Trader Joe's – нет. Тем не менее, здесь мы можем применить деятельность исходящей логистики, чтобы обозначить ряд удобств, с которыми покупатели сталкиваются, когда они находятся в магазине Trader Joe's. Компания тщательно продумала, какой опыт она хочет, чтобы мы получили, посещая ее магазины.
Среди многих тактических логистических операций Trader Joe есть дегустации в магазине. Обычно одновременно проводится несколько дегустаций продуктов, которые создают живую атмосферу и часто совпадают с сезонами и праздниками. На дегустационных станциях представлены как новые, так и знакомые блюда, которые готовят и обслуживают сотрудники.
4. Маркетинг и продажи. По сравнению со своими конкурентами Trader Joe's почти не занимается традиционным маркетингом. Тем не менее, весь его опыт в магазине является формой маркетинга. Копирайтеры компании создают этикетки для продуктов специально для своей клиентской базы. Уникальный брендинг и инновационная культура Trader Joe's указывают на то, что компания хорошо знает своих клиентов, что и должно быть, поскольку фирма действительно выбрала тот тип клиентов, который ей нравится, и не отклонилась от этой модели.
Благодаря этому непрямому маркетингу стиля и имиджа Trader Joe's удалось выделиться на рынке, тем самым укрепив свое конкурентное преимущество.
5. Сервис. Обслуживание клиентов имеет первостепенное значение для Trader Joe's. Как правило, вы видите в два раза больше сотрудников, чем покупателей в их магазинах. Какой бы работой они ни занимались в данный момент, дружелюбный, знающий и красноречивый персонал всегда готов помочь вам . Сотрудники приветствуют вмешательство покупателей и немедленно спешат найти ваш товар или ответить на ваш вопрос. Кроме того, компания всегда использовала программу возврата без вопросов. Не понравится — вернешь деньги — и точка.
Этот список можно продолжать и продолжать, прежде чем он дойдет до четырех видов поддержки, упомянутых выше, поскольку Trader Joe's является чрезвычайно успешным примером применения теории цепочки создания стоимости в своем бизнесе.
химический элемент | Определение, происхождение, распространение и факты
химический элемент
Посмотреть все СМИ
- Ключевые люди:
- Лотар Мейер Антуан Лавуазье Дмитрий Менделеев Луи Бернар Гайтон де Морво Джозеф Лошмидт
- Связанные темы:
- редкоземельный элемент изотоп переходный металл периодическая таблица водород
Просмотреть весь соответствующий контент →
Резюме
Прочтите краткий обзор этой темы
химический элемент , также называемый элемент , любое вещество, которое не может быть разложено на более простые вещества с помощью обычных химических процессов. Элементы — это основные материалы, из которых состоит вся материя.
В этой статье рассматривается происхождение элементов и их изобилие во Вселенной. Подробно рассмотрено геохимическое распространение этих элементарных веществ в земной коре и недрах, их нахождение в гидросфере и атмосфере. В статье также рассматривается периодический закон и основанное на нем табличное расположение элементов. Для получения подробной информации о соединениях элементов см. Химическое соединение .
Редакция Британской энциклопедии
Общие наблюдения
В настоящее время известно 118 химических элементов. Около 20% из них не существуют в природе (или присутствуют лишь в следовых количествах) и известны только потому, что получены синтетическим путем в лаборатории. Из известных элементов 11 (водород, азот, кислород, фтор, хлор и шесть инертных газов) являются газами при обычных условиях, два (бром и ртуть) являются жидкостями (еще два, цезий и галлий, плавятся при температуре около или около выше комнатной температуры), а остальные – твердые вещества. Элементы могут соединяться друг с другом, образуя широкий спектр более сложных веществ, называемых соединениями. Количество возможных соединений почти бесконечно; известно, возможно, миллион, и каждый день открываются новые. Когда два или более элемента объединяются, образуя соединение, они теряют свою самостоятельную идентичность, и продукт приобретает характеристики, совершенно отличные от характеристик составляющих его элементов. Газообразные элементы водород и кислород, например, с совершенно разными свойствами, могут соединяться, образуя сложную воду, свойства которой совершенно отличаются от кислорода или водорода. Вода явно не является элементом, потому что она состоит из двух веществ, водорода и кислорода, и может быть химически разложена на них; эти два вещества, однако, являются элементами, потому что они не могут быть разложены на более простые вещества ни одним известным химическим процессом. Большинство образцов природного вещества представляют собой физические смеси соединений. Морская вода, например, представляет собой смесь воды и большого количества других соединений, наиболее распространенным из которых является хлорид натрия, или поваренная соль. Смеси отличаются от соединений тем, что их можно разделить на составные части с помощью физических процессов; например, простой процесс испарения отделяет воду от других соединений в морской воде.
Историческое развитие концепции элемента
Современная концепция элемента недвусмысленна, поскольку она зависит от использования химических и физических процессов в качестве средства различения элементов от соединений и смесей. Однако существование фундаментальных субстанций, из которых состоит вся материя, было основой многих теоретических предположений с самого начала истории. Древнегреческие философы Фалес, Анаксимен и Гераклит предполагали, что вся материя состоит из одного существенного начала — или элемента. Фалес считал, что этим элементом является вода; Анаксимен предложил воздух; и Гераклит, огонь. Другой греческий философ, Эмпедокл, высказывал другое мнение, что все вещества состоят из четырех элементов: воздуха, земли, огня и воды. Аристотель соглашался и подчеркивал, что эти четыре элемента являются носителями основных свойств: сухость и тепло связаны с огнем, тепло и влага с воздухом, влага и холод с водой, холод и сухость с землей. В мышлении этих философов предполагалось, что все остальные субстанции представляют собой комбинации четырех элементов, а свойства субстанций отражали их составы элементов. Таким образом, греческая мысль заключала в себе идею о том, что вся материя может быть понята с точки зрения элементарных качеств; в этом смысле сами элементы мыслились как нематериальные. Греческое понятие элемента, которое было принято почти 2000 лет, содержало только один аспект современного определения, а именно, что элементы обладают характерными свойствами.
Britannica Quiz
Science Quiz
Проверь свои научные способности под микроскопом и узнай, что ты знаешь о кровавых камнях, биомах, плавучести и многом другом!
В конце Средневековья, когда алхимики стали более изощренными в своих знаниях о химических процессах, греческие представления о составе материи стали менее удовлетворительными. Дополнительные свойства элементов были введены для учета вновь открытых химических превращений. Таким образом, сера стала представлять качество горючести, ртуть — летучести или текучести, а соль — устойчивости к огню (или негорючести). Эти три алхимических элемента или принципа также представляли собой абстракции свойств, отражающие природу материи, а не физических субстанций.
В конце концов была понята важная разница между смесью и химическим соединением, и в 1661 году английский химик Роберт Бойль признал фундаментальную природу химического элемента. Он утверждал, что четыре греческих элемента не могут быть настоящими химическими элементами, потому что они не могут соединяться с образованием других веществ и не могут быть извлечены из других веществ. Бойль подчеркивал физическую природу элементов и связывал их с соединениями, которые они образовывали современным оперативным путем.
Оформите подписку Britannica Premium и получите доступ к эксклюзивному контенту. Подпишитесь сейчас
В 1789 году французский химик Антуан-Лоран Лавуазье опубликовал то, что можно считать первым списком элементарных веществ, основанным на определении Бойля. Список элементов Лавуазье был составлен на основе тщательного количественного изучения реакций разложения и рекомбинации. Поскольку он не мог разработать опыты по разложению некоторых веществ или их образованию из известных элементов, Лавуазье включил в свой список элементов такие вещества, как известь, оксид алюминия и кремнезем, которые теперь известны как очень устойчивые соединения. На то, что Лавуазье все еще сохранил определенное влияние древнегреческой концепции элементов, указывает его включение света и тепла (калорийных) в число элементов.
Семь веществ, признанных сегодня элементами — золото, серебро, медь, железо, свинец, олово и ртуть — были известны древним, потому что они встречаются в природе в относительно чистом виде. Они упоминаются в Библии и в раннем индуистском медицинском трактате Чарака-самхита . Шестнадцать других элементов были открыты во второй половине 18 века, когда стали лучше понятны методы отделения элементов от их соединений. Еще восемьдесят два последовали за введением методов количественного анализа.