Последовательность | музыкальная композиция | Британика
последовательность
Просмотреть все носители
- Связанные темы:
- музыкальная форма
Просмотреть весь связанный контент →
последовательность , в музыке мелодическая или аккордовая фигура, повторяющаяся на новом уровне высоты тона (т. е. транспонированная), таким образом объединяющая и развивающая музыкальный материал. Слово последовательность имеет два основных значения: средневековая последовательность в литургии латинской мессы и гармоническая последовательность в тональной музыке.
В средневековой музыке и литературе последовательность представляла собой латинский текст, связанный с определенной напевной мелодией, которую пели на мессе между Аллилуйей и чтением Евангелия. Он развился примерно в 9 веке от тропа (добавление музыки, текста или того и другого) до jubilus , витиеватого окончания последнего слога Аллилуйи.
К XI веку последовательность приобрела общую поэтическую форму, отражающую музыкальную структуру: обычно вступительные и заключительные строки заключали в себе серию рифмованных метрических куплетов разной длины ( x aa bb cc … у ). Каждому слогу соответствовала одна музыкальная нота. В конце концов, тексты были установлены на вновь сочиненные мелодии, а длина куплетов была уравнена. Последовательности стали очень популярными по всей Европе, и сохранились тысячи их примеров, подходящих для различных литургических праздников. В 16 веке Тридентский собор отменил все последовательности литургии, кроме четырех: Victimae paschali laudes («Хвала Пасхальной Жертве»), Veni Sancte Spiritus («Приди Святый Дух»), Lauda Sion («Хвала Сиону») и D («День гнева»). Stabat mater dolorosa («Скорбящая мать стояла») был восстановлен в 1727 году.0017 lai (жанр песни труверов, средневековых французских поэтов-композиторов).
Stabat mater dolorosa («Скорбящая мать стояла») был восстановлен в 1727 году.0017 lai (жанр песни труверов, средневековых французских поэтов-композиторов).
В тональной музыке гармоническая последовательность, как аккомпанемент для мелодии, представляет собой мотивный паттерн из двух или более последовательных гармоний, который воспроизводится в транспозиции, как правило, два или три раза, сохраняя одну и ту же мелодическую форму (относительное движение) каждой часть или голос. Создавая гармоническое и тональное разнообразие с единым рисунком, последовательность служит средством музыкального развития. Обычно используются два типа последовательности:
Гармоническая последовательность, хотя ею легко злоупотреблять при механическом применении, широко использовалась всеми композиторами тональной музыки, то есть теми, кто работал примерно с 1700 по 1900 год.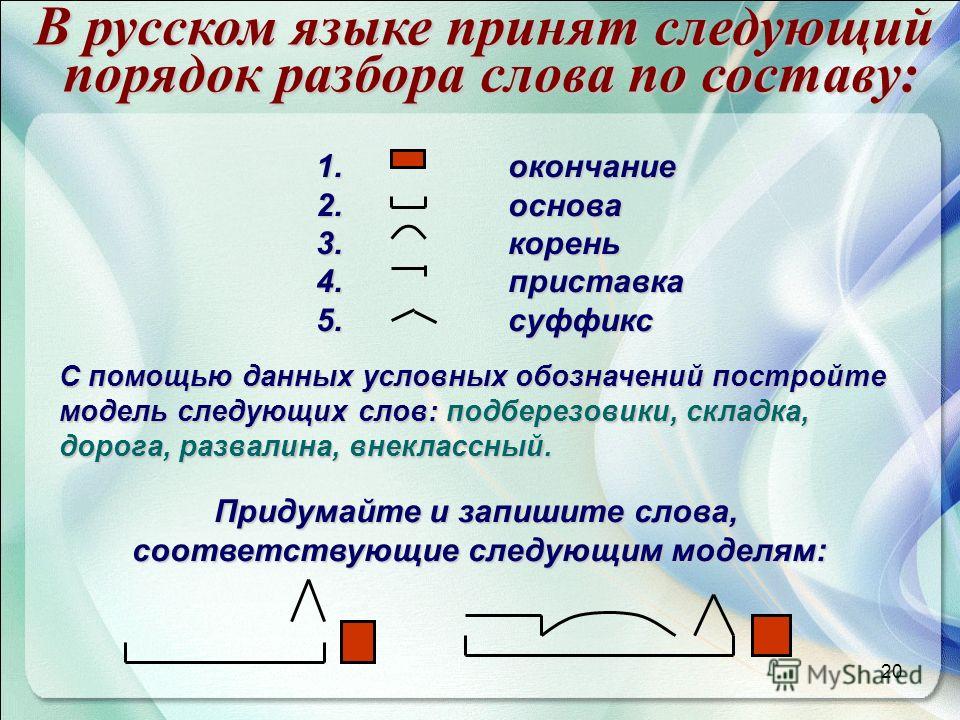 Очень длинные последовательности появляются в концертах эпохи барокко, особенно в произведениях Джорджа Фридриха Генделя и Антонио Вивальди. Часто последовательность используется для модуляции в развивающей части сонатной формы, как в первой части бетховенской 9-й0017 Симфония № 1 до мажор (18:00). Замечательная расширенная серия модулирующих секвенций характерна для развивающей части Концерта для фортепиано с оркестром № 1 ми минор
Очень длинные последовательности появляются в концертах эпохи барокко, особенно в произведениях Джорджа Фридриха Генделя и Антонио Вивальди. Часто последовательность используется для модуляции в развивающей части сонатной формы, как в первой части бетховенской 9-й0017 Симфония № 1 до мажор (18:00). Замечательная расширенная серия модулирующих секвенций характерна для развивающей части Концерта для фортепиано с оркестром № 1 ми минор
Оформите подписку Britannica Premium и получите доступ к эксклюзивному контенту. Подпишитесь сейчас
Марк ДеВото
Сравнение последовательностей – My Biosoftware – Блог о программном обеспечении для биоинформатики
:: ОПИСАНИЕ
Spaced Words – это новый подход к сравнению последовательностей без выравнивания. В то время как большинство алгоритмов без выравнивания сравнивают словесный состав последовательностей, интервальные слова используют шаблон «заботливые» и «безразличные» позиции.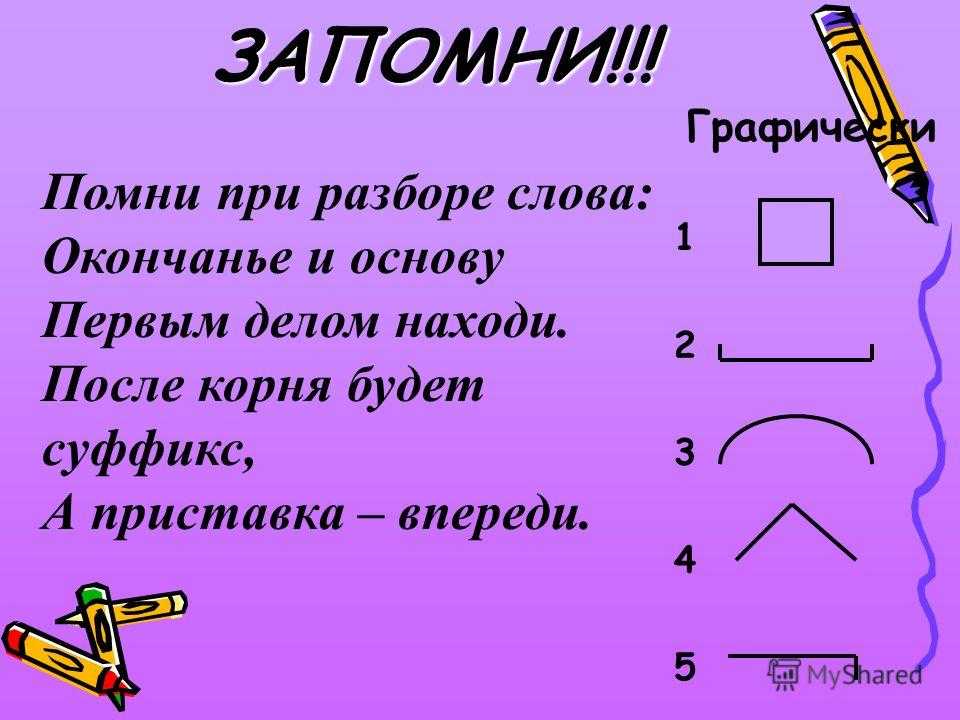
:: Разработчик
Департамент биоинформатики, Университет Гёттингена
:: Скриншоты
N/A
:: требования
- :: Требования
- Linux/MacOsX
- Компилятор С++
- Linux/MacOSX/Windows
- Р
- Linux /Mac 9 OsX0 / Windows
- Linux / macosx / Window
- Ява
- Linux
- Web Browsher :: Требования
- Linux ::
- Компилятор С++
- Windows / Linux / MacOSX
- Компилятор С++
:: СКАЧАТЬ
Spaced Words
:: ПОДРОБНЕЕ
Цитирование
Биоинформатика. 3 апреля 2014 г.
Быстрое сравнение последовательностей без выравнивания с использованием частот разделенных слов.
Леймейстер СА1, Боден М., Хорвеге С., Линднер С., Моргенштерн Б.
:: ОПИСАНИЕ
D2NGS — это пакет C++, предназначенный для реализации программы для сравнения последовательностей генома без выравнивания с использованием данных NGS с использованием d2, d2* и d2S.
::РАЗРАБОТЧИК
Fengzhu Sun
:: СКРИНШОТЫ
Н/Д
:: ТРЕБОВАНИЯ
:: Скачать
D2NGS
:: Дополнительная информация
Цитация
Song K, Ren J, Zhai ZY, Liu XM, Deng MH и Sun FZ (2012)
Сравнение последовательности. На основе чтения секвенирования следующего поколения: расширенный реферат
Research in Computational Molecular Biology, 272-285.
:: ОПИСАНИЕ
multiAlignFree — пакет R, предназначенный для реализации программы для множественного выравнивания без сравнение последовательностей на основе длинной последовательности генома или данных NGS.
::РАЗРАБОТЧИК
Fengzhu Sun
:: СКРИНШОТЫ
Н/Д
:: ТРЕБОВАНИЯ
:: СКАЧАТЬ
multiAlignFree
:: ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Цитирование
Биоинформатика. 2013 1 ноября; 29 (21): 2690-8. doi: 10.1093/биоинформатика/btt462. Epub 2013, 29 августа.
Множественное сравнение последовательностей без выравнивания.
Рен Дж.1, Сонг К., Сун Ф., Дэн М., Райнерт Г.
:: ОПИСАНИЕ
Программы FASTA находят области локального или глобального (нового) сходства между последовательностями белков или ДНК либо путем поиска в базах данных белков или ДНК, либо путем выявления локальных дубликатов в последовательности.
::РАЗРАБОТЧИК
Уильям Р. Пирсон
:: СКРИНШОТЫ
Н/Д
:: ТРЕБОВАНИЯ
:: СКАЧАТЬ
FASTA
:: ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Цитирование:
Pearson WR, Lipman DJ.
Улучшенные инструменты для сравнения биологических последовательностей. Proc Natl Acad Sci U S A. 1988 Apr; 85(8):2444-8.
 3.8h – Сравнение последовательностей
3.8h – Сравнение последовательностей:: ОПИСАНИЕ разработать новый инструмент визуализации, позволяющий проводить эффективный сравнительный анализ последовательности генома. Программа будет сравнивать несколько последовательностей из предположительно гомологичных областей у разных видов. Результаты различных существующих программ анализа, таких как программы прогнозирования генов, гомологии белков и программ прогнозирования регуляторных последовательностей, должны быть визуализированы и использованы для поиска соответствующих доменов последовательностей.
: : Developer
Niclas Jareborg
:: Скриншоты
:: Требования
:: СКАЧАТЬ
Alfresco
:: ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Citation
Alfresco – инструмент для сравнительного анализа геномных последовательностей.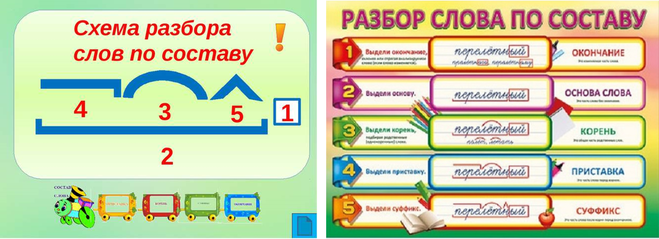
Jareborg N, Durbin R.
Genome Res. 2000 авг.; 10(8):1148-57.
:: ОПИСАНИЕ is new a a
km km::РАЗРАБОТЧИК
Факультет биоинформатики, Геттингенский университет
:: СКРИНШОТЫ
Н/Д
:: ТРЕБОВАНИЯ
:: СКАЧАТЬ
kmacs
:: ПОДРОБНЕЕ
Цитирование
Биоинформатика. 2014 13 мая. pii: btu331. [Epub перед печатью]
2014 13 мая. pii: btu331. [Epub перед печатью]
kmacs: подход k-Mismatch Average Common Substring к сравнению последовательностей без выравнивания.
Леймейстер CA1, Моргенштерн B2.
:: ОПИСАНИЕ
AnABlast представляет собой алгоритм для обнаружения сигналов последовательностей, кодирующих белок, в областях генома. Вы можете анализировать короткую последовательность нуклеотидов (длиной до 25 КБ или до 1 МБ, если вы загружаете отчет Blast). Он выделяет геномные области с наложенными друг на друга незначительными выравниваниями (протомотивами), которые представляют существующие или древние последовательности, кодирующие белок. Он позволяет открывать новые гены у бактерий или экзоны у эукариотических организмов.
:: Developer
Вычислительная биология и анализ данных (CBDM) Группа
:: Скриншоты
N/A
:: Требования
- :: Требования
:: СКАЧАТЬ
NO
:: ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Citation
Methods Mol Biol. 2019; 1962: 207-214. дои: 10.1007/978-1-4939-9173-0_12.
2019; 1962: 207-214. дои: 10.1007/978-1-4939-9173-0_12.
AnABlast: повторный поиск кодирующих белок последовательностей в геномных регионах.
Рубио А., Касимиро-Соригер С.С., Миер П., Андраде-Наварро М.А., Гарсон А., Хименес Х., Перес-Пулидо А.Дж.
:: ОПИСАНИЕ
LAST обеспечивает быстрое и точное сравнение больших последовательностей произвольно неоднородного состава. LAST похож на BLAST, но лучше справляется с биологическими последовательностями гигамасштаба. Он также может использовать данные о качестве последовательности и указывать неоднозначность каждого столбца в выравнивании.
:: Developer
Исследовательский центр вычислительной биологии [CBRC]
:: Скриншоты
N/A
:: Требования
- ::
:: СКАЧАТЬ
ПОСЛЕДНЯЯ
:: ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Citation
Адаптивные семена приручают сравнение геномной последовательности.
С. М. Килбаса, Р. Ван, К. Сато, П. Хортон, М. С. Фрит,
Рез. генома. 2011 март; 21(3):487-93. doi: 10.1101/гр.113985.110
:: DESCRIPTION
9000 эффективный метод крупномасштабного сравнения последовательностей, использующий позиционное хеширование.::РАЗРАБОТЧИК
Исследовательская лаборатория биоинформатики, Медицинский колледж Бейлора
:: СКРИНШОТЫ
Н/Д
:: ТРЕБОВАНИЯ
:: Скачать
Pash
:: Дополнительная информация
Citation
Coarfa C, Yu F, Miller CA, Chen Z, Harris RA, Milosavljevic A.